
Трисомия 18 хромосомы что это – Синдром Эдвардса (трисомия 18). Причины, симптомы, признаки, диагностика и лечение патологии :: Polismed.com
Синдром Эдвардса — Википедия
Материал из Википедии — свободной энциклопедии
Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 25 мая 2015; проверки требуют 16 правок. Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 25 мая 2015; проверки требуют 16 правок.Синдром Э́двардса (синдром трисомии 18) — хромосомное заболевание, характеризуется комплексом множественных пороков развития и трисомией 18 хромосомы. Описан в 1960 году Джоном Эдвардсом (John H. Edwards). Популяционная частота примерно 1:3000 в США, и 1:5000 в мире на 2016 год. Дети с трисомией в 18 хромосоме чаще рождаются у пожилых матерей, взаимосвязь с возрастом матери менее выражена, чем в случаях трисомии хромосомы 21[3] и 13[4]. Для женщин старше 45 лет риск родить больного ребёнка составляет 0,7 %. Девочки с синдромом Эдвардса рождаются в три раза чаще мальчиков. Выживание после года жизни составляет около 5–10%
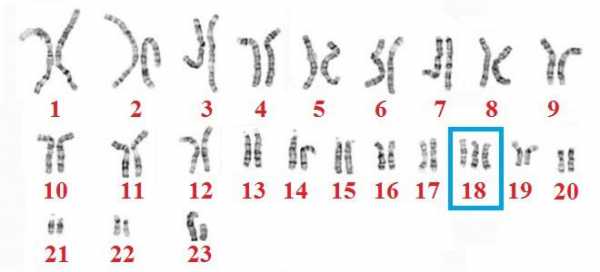
Причиной заболевания является наличие дополнительной 18-й хромосомы (трёх вместо двух в норме для диплоидного набора) в кариотипе зиготы.
Лишняя хромосома обычно появляется до оплодотворения. У человека нормальные половые клетки — гаметы — содержат по 23 хромосомы (гаплоидный набор) и, сливаясь, они дают кариотип зиготы — 46 хромосом. К появлению лишней хромосомы у гамет обычно приводит нерасхождение хромосом при мейотическом делении, вследствие чего в половой клетке оказывается 24 хромосомы. В случае, если такая клетка встретит при оплодотворении гамету от противоположного пола, они образуют зиготу с трисомией.
В одном случае из десяти наблюдается мозаицизм в явлении трисомии 18: лишнюю хромосому несут не все клетки организма. Это говорит о том, что нерасхождение произошло на ранней стадии развития зародыша, а все клетки с трисомией — потомки неправильно поделившейся клетки зародыша.
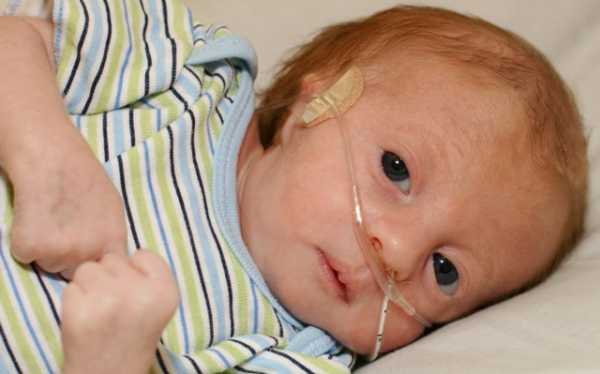
Дети с трисомией 18 рождаются с низким весом, в среднем около 2200 грамм, при этом длительность беременности — нормальная или даже превышает норму. Фенотипические проявления синдрома Эдвардса многообразны. Чаще всего возникают аномалии мозгового и лицевого черепа, мозговой череп имеет долихоцефалическую форму. Нижняя челюсть и ротовое отверстие маленькие. Глазные щели узкие и короткие. Ушные раковины деформированы и в подавляющем большинстве случаев расположены низко, несколько вытянуты в горизонтальной плоскости. Мочка, а часто и козелок отсутствуют. Наружный слуховой проход сужен, иногда отсутствует. Грудина короткая, из-за чего межреберные промежутки уменьшены и грудная клетка шире и короче нормальной. В 80 % случаев наблюдается аномальное развитие стопы: пятка резко выступает, свод провисает (стопа-качалка), большой палец утолщён и укорочен. Из дефектов внутренних органов наиболее часто отмечаются пороки сердца и крупных сосудов: дефект межжелудочковой перегородки, аплазии одной створки клапанов аорты и лёгочной артерии. У всех больных наблюдаются гипоплазия мозжечка и мозолистого тела, изменения структур олив, выраженная умственная отсталость, снижение мышечного тонуса, переходящее в повышение со спастикой.
Продолжительность жизни детей с синдромом Эдвардса невелика: 60 % детей умирают в возрасте до 3 месяцев, до года доживает лишь 5-10 %. Основной причиной смерти служат остановка дыхания и нарушения работы сердца. Оставшиеся в живых — глубокие олигофрены.
Частота появления синдрома Эдвардса составляет ~ 1:7000 зачатий и 1:8000 рождений живых детей. Риск рождения больного ребёнка увеличивается с возрастом, особенно, если мать болеет диабетом.
Кроме трисомии 18, присутствующей во всех клетках организма, а также мозаичной трисомии 18, возможна и частичная трисомия. При этом часть хромосомы 18 присоединяется к другой хромосоме. Такой эффект называется транслокация, и он может произойти как при созревании гамет, так и после оплодотворения в клетках зародыша. В клетках организма при этом оказываются две гомологичные хромосомы 18 и, дополнительно, часть хромосомы 18, прикреплённая к другой хромосоме. У людей, страдающих частичной трисомией 18, аномалии проявляются слабее, нежели при типичном синдроме Эдвардса.
ru.wikipedia.org
Синдром Эдвардса: причины, признаки, диагностика, лечение
Синдром Эдвардса – заболевание, обусловленное спонтанной мутацией генов и появлением дополнительной 18 хромосомы. Подобные патологические изменения происходят в организме плода во время эмбриогенеза. Трисомия по 18-ой аутосоме сопровождается разнообразными пороками развития, приводящими к инвалидности или смерти ребенка. Синдром Эдвардса распространен по всему земному шару без четкой зависимости от местности или расы.
У больных детей изменяется внешний вид, поражается опорно-двигательная, пищеварительная, сердечно-сосудистая, нервная и мочеполовая системы. Следствием количественной хромосомной аберрации является своеобразный фенотип — длинная голова; недоразвитые уши, глаза и челюсти; короткая верхняя губа; косолапость. Синдром Эдвардса проявляется глубокой умственной отсталостью и многочисленными врожденными пороками внутренних органов: сердца, мозга, почек.
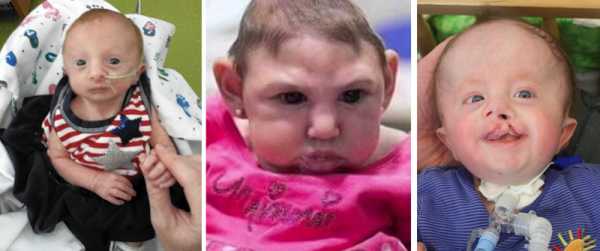
дети с синдромом Эдвардса
Впервые патологию описал ученый-генетик J. Edwards в 1960 году. Он выделил более 100 симптомов заболевания. Благодаря этому открытию синдром получил свое название. Синдром Эдвардса по распространенности среди хромосомных недугов уступает только синдрому Дауна. Трисомия – вариант хромосомной мутации, при которой у человека в клетках содержится не 46, а 47 хромосом. Существует всего 3 синдрома в данной группе нозологий: синдром Эдвардса (трисомия 18 хромосомы), Дауна (трисомия 21 хромосомы) и Патау (трисомия 13 хромосомы). При наличии других добавочных хромосом заболевание несовместимо с жизнью.
Согласно статистики, болеют синдромом Эдвардса преимущественно девочки. Женщины, носящие под сердцем плод мужского пола, обычно не вынашивают беременность – она заканчивается самопроизвольным абортом. Риск развития синдрома возрастает в случае беременности после 40 лет. Болезнь негативно сказывается на течении беременности: отмечается многоводие, недостаточная активность плода и поздние роды – после 42 недель.
Диагностика синдрома состоит из трех этапов — до зачатия, до родов и после рождения ребенка. Проводят ультразвуковое и цитогенетическое исследования плода, а также используют инвазивные методы, позволяющие выявить недуг пренатально. Новорожденных детей осматривают специалисты в области неонтологии, детской кардиологии, неврологии, хирургии, ортопедии, урологии. Им делают ЭКГ, УЗИ почек, исследование органов брюшной полости. Синдром Эдвардса — генетическое заболевание, вылечить которое невозможно. Больным детям требуется симптоматическая терапия и тщательный уход. Современные методы лечения способны поддержать жизнь ребенка на оптимальном уровне и добиться определенного прогресса в его развитии.
Большинство детей с синдромом Эдвардса погибают внутриутробно. Новорожденные девочки редко доживают до 8-10 месяцев, а мальчики — до 2-3- месяцев. Только 10% рожденных детей способны прожить год. Взрослыми становятся лишь единицы. Больные дети умирают от сердечной дисфункции, воспаления легких, удушья, кишечной непроходимости. Эти осложнения обусловлены врожденные пороками развития.
Синдром Эдвардса – показание к прерыванию беременности. Рождение больных деток является осознанным выбором родителей. Чаще всего так происходит у женщин, которые не становились на учет по беременности и не проходили рекомендуемых исследований. Обычно родители отказываются от больного ребенка, практически обреченного на гибель.
Причины
Кариотип здорового ребенка состоит из 46 хромосом: по 23 от обоих родителей. У лиц с синдромом Эдвардса под влиянием не установленных наукой факторов происходит дублирование генетического материала, и появляется дополнительная 47 хромосома, которая является «лишней». Обычно мутации подвергается 18 хромосома. Так формируется название – трисомия 18. Хромосомные нарушения происходят в процессе образования гамет или дробления зиготы. В большинстве случаев возникает простая трисомия 18, крайне редко – мозаичная или транслокационная форма. Причины синдрома Эдвардса в настоящее время остаются неизвестными. Больной ребенок может появиться на свет в семье, где родители и родственники являются абсолютно здоровыми.
- Полная трисомия – три 18 хромосомы в каждой клетке плода. Патология обусловлена нерасхождением хромосом в процессе мейоза. Почти всегда лишняя хромосома передается по материнской линии. Этот вариант синдрома Эдвардса протекает довольно тяжело по сравнению с другими формами, встречается намного чаще и практически всегда имеет неблагоприятный прогноз.
- Мозаицизм связан с нерасхождением хромосом после слияния половых клеток на ранней стадии дробления зиготы. Обе гаметы изначально имеют нормальный набор хромосом, но в результате удвоения генетического материала и формирования зародыша происходит сбой. При этом только часть клеток плода получит лишнюю хромосому. Доля патологических клеток никогда не превышает 50%. Их число зависит от того, на каком этапе деления начальной клетки произошел сбой. Чем позже это происходит, тем меньше будет доля дефектных клеток. Общее состояние пациента при этом легче, чем при классической форме трисомии 18.
- Частичная трисомия или транслокация – добавление фрагмента третьей хромосомы в результате дефекта деления генетического материала. Транслокационная перестройка ведет к избыточности информации и нарушению генетической последовательности в двух хромосомах. Гены 18 хромосомы переходят с одного участка на другой. Для пациентов с частичной трисомией 18 прогноз лучше, чем для детей с полной формой, но все равно остается неблагоприятным.
Клинически все 3 варианта данного синдрома протекают по одному типу, но первый вариант все же может отличаться более тяжелой формой.
Факторы, способствующие развитию патологии:
- неблагоприятная экология,
- облучение,
- воздействие химикатов и прочих токсинов,
- прием алкоголя беременной женщиной,
- активное и пассивное курение,
- воздействие некоторых лекарств,
- кровное родство супругов,
- заболевания половой сферы,
- возраст матери старше 40 лет.
Вышеперечисленные факторы лишь повышают риск развития данной мутации, а не являются ее непосредственными причинами.
Передача измененного набора хромосом последующим поколениям невозможна. Большинство больных не доживают до репродуктивного возраста. Репродуктивные органы, как и репродуктивные способности у них недоразвиты. Синдром Эдвардса не передается по наследству.
Симптомы
Избыток генетической информации в клетках приводит к появлению соответствующих симптомов болезни, которые объединены под названием «синдром Эдвардса».
Первые признаки патологии появляются во время беременности:
- Многоводие,
- Слабая активность плода,
- Гипоплазия плаценты,
- Аномальное строение пуповины.
Новорожденные дети имеют низкую массу тела – около 2 кг, а также признаки гипотрофии при доношенной или переношенной беременности. У них нарушен процесс дыхания, сосания и проглатывания молока. Им требуется питание через зонд и длительная вентиляция легких.
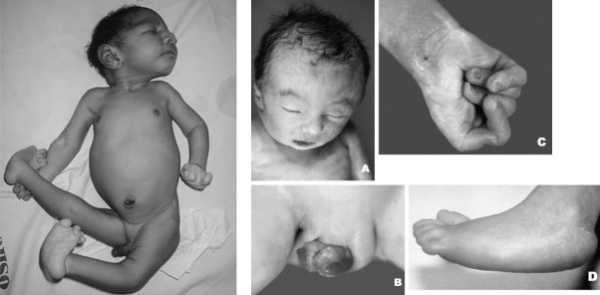
Фенотипические признаки новорожденных с синдромом Эдвардса:
- Долихоцефалия — длинноголовость,
- Непропорционально маленькая голова,
- Низкий лоб,
- Выступающий затылок,
- «Готическое» небо,
- Узкие глазные щели,
- Узкий и вдавленный нос,
- Микрогнатия — маленькая челюсть,
- Микрофтальмия — мелкие глазные яблоки,
- «Заячья губа» и «волчья пасть»,
- Вертикальная складка на внутреннем уголке глаза,
- Патологически неправильный прикус,
- Искаженные черты лица,
- Опущение верхнего века,
- Стробизм,
- Короткая шея,
- Деформация ушных раковин,
- Низкая посадка ушей.
Характерные деформации скелета:
- синдактилия кистей,
- укороченная и расширенная грудина,
- косолапость,
- искривление позвоночника,
- гипо- и атрофия мышц,
- «стопа-качалка»,
- полное слияние или перепончатость пальцев на нижних конечностях,
- аномальное сгибание и разгибание суставов,
- флексорное положение кисти.
Тяжелые аномалии со стороны внутренних органов:
- врожденные пороки сердца,
- грыжи,
- меккелев дивертикул,
- сужение привратника,
- отсутствие анального отверстия,
- удвоение мочеточника,
- почка в форме подковы,
- расширение чашечно-лоханочного комплекса,
- выпячивание стенки мочевого пузыря,
- неопущение яичка в мошонку,
- двурогая матка,
- клиторомегалия,
- атрофия или сглаживание извилин мозга,
- микроцефалия,
- менингомиелоцеле,
- водянка головного мозга,
- субарахноидальные кисты,
- недоразвитие мозжечка и мозолистого тела.
Нарушения психической сферы:
- умственная отсталость,
- олигофренизм,
- имбицилизм,
- идиотизм,
- задержка нервно-психического развития.
Тяжелейшие пороки фактически не дают больным шанса на выживание. Даже качественное лечение не спасает детей от гибели на первом году жизни.
Перечисленные клинические признаки синдрома Эдвардса довольно специфичны и разнообразны. Они позволяют заподозрить у пациента данный недуг и поставить предварительной диагноз. Сочетание наиболее частых симптомов с высокой вероятностью говорит о наличии у ребенка тяжелой патологии.
Диагностика
Поскольку синдром Эдвардса характеризуется довольно большим количеством ярко выраженных отклонений, его довольно просто диагностировать даже по внешним проявлениям. Однако этого недостаточно, чтобы поставить окончательный диагноз.
Диагностика синдрома Эдвардса складывается из трех этапов — обследование супружеских пар до момента зачатия, беременной женщины до родов и ребенка после появления на свет.
Диагностика до зачатия ребенка — идеальный вариант, но не всегда применимый. Специалисты-генетики могут лишь предположить, каков риск рождения ребенка с хромосомным заболеванием в данной семье.
- До момента зачатия врачи собирают семейный анамнез, опрашивая родителей об их родословной.
- Большое внимание специалисты уделяют факторам риска: возрасту матери, перенесенным инфекционным заболеваниям, хроническим болезням, вредным привычкам.
- Генетический анализ родителей – полноценное исследование, с помощью которого составляется их кариотип и обнаруживаются участки ДНК с дефектными генами.
Диагностика в период внутриутробного развития дает более точные результаты, поскольку обследуют организм плода. Пренатальная диагностика — важный этап в процессе выявления хромосомных нарушений.
- Ультразвуковое исследование плода и допплерография маточно-плацентарного кровотока – неинвазивные методы, полностью безопасные и рекомендованные всем беременным. Признаки синдрома Эдвардса: отставание плода в размерах и массе, большое количество околоплодных вод, видимые аномалии развития черепа и костей, агенезия пупочной артерии, малая величина плаценты, многоводие, брадикардия, отсутствие носовых костей, 2 артерии в пуповине, кисты сосудистых сплетений. Диагностика с помощью ультразвукового исследования является достоверной на 100%.
- Стандартный пренатальный скрининг включает анализ крови на сывороточные маркеры. Полученные результаты соотносят с возрастом беременной женщины и сроком гестации. При отклонении показателей от нормы ставят высокий риск синдрома Эдвардса. В таких случаях показано искусственное прерывание беременности по медицинским показаниям.
- Амниоцентез – клеточный анализ околоплодных вод. Инвазивная методика, осуществляемая путем забора амниотической жидкости шприцем. Ее клетки содержат образцы ДНК плода, которые проверяют на наличие генетических заболеваний.
- Кордоцентез — исследование пупочной крови плода, позволяющее определить генетические аномалии с высокой точностью.
- Биопсия хориона представляет собой пункцию матки через переднюю брюшную стенку и забор ткани для анализа – стандартного генетического исследования.
Беременным, попадающим в группу высокого риска, предлагается проведение инвазивной дородовой диагностики с последующим кариотипированием плода. Инвазивные методы считаются самыми точными и надежными, но требующими оперативного вмешательства и проникновения в оболочку плода. Диагноз подтверждается при помощи определения кариотипа малыша путем КФ-ПЦР.
Диагностика синдрома Эдвардса после рождения самая легкая, быстрая и точная. После выявления некоторых врожденных дефектов проводят генетический анализ для подтверждения диагноза. Основной задачей при рождении ребенка с этой патологией является обнаружение аномалий в развитии внутренних органов, которые обычно приводят к смерти в первые месяцы жизни. Именно на их поиск направлено большинство диагностических процедур непосредственно после рождения. Наиболее важными диагностическими исследованиями, которые должны быть выполнены ребенку с синдромом Эдвардса в первые часы жизни, являются эхокардиография, УЗИ органов брюшной полости и УЗИ почек.
Видео: синдром Эдвардса на УЗИ у плода, расширение воротниковой зоны
Лечение
Синдром Эдвардса, как и все хромосомные аномалии, неизлечим. Патология в большинстве случаев несовместима с жизнью. Больным детям показана симптоматическая терапия, направленная на поддержание физиологических функций организма, продление жизни и улучшение ее качества.
Коррекция патологических процессов, опасных для жизни, заключается в назначении противовоспалительного и антибактериального лечения при наличии пневмонии, восстановлении проходимости пищеварительного тракта при атрезии кишечника, кормлении через зонд, использовании пеногасителей при метеоризме, слабительных средств при запоре. Для поддержания жизнеспособности всего организма проводится коррекция работы дыхательной и пищеварительной систем, а также нормализация сердечной деятельности. Больным необходимо создать стерильные условия, чтобы избежать развития различных инфекционных заболеваний. Дети с данным синдромом требуют тщательного ухода и регулярного прохождения медицинского обследования.
Пупочные и паховые грыжи, пороки сердца и прочие аномалии лечат хирургическим путем, если состояние больного ребенка остается удовлетворительным. Оперативное вмешательство несет неоправданный риск. Хирурги, исправив внешние дефекты, могут потерять пациента из-за сбоя в работе сердечно-сосудистой системы и развития прочих серьезных осложнений.
Все дети с синдромом Эдвардса без исключения имеют отклонения в психофизическом развитии. Им требуется специальная программа воспитания, включающая комплекс обучающих методик. Она не восстановит нарушенные функции, но поможет привить некоторые элементарные бытовые навыки.
Прогноз и профилактика
Прогноз при синдроме Эдвардса крайне неблагоприятный. Больные дети погибают на первом году жизни. Только единицы доживают до юных и зрелых лет. При этом они имеют значительные умственные отклонения и нуждаются в постоянном уходе. Выжившие дети страдают от умственной отсталости и различных заболеваний, связанных с аномалиями строения. Только легкая форма мозаичного синдрома Эдвардса поддается коррекции. Больные дети начинают общаться с ограниченным кругом людей и приобретают элементарные навыки самообслуживания. Если обеспечить необходимый уход, ребенок может научиться самостоятельно поднимать голову и есть.
Чтобы предотвратить рождение больного ребенка, необходимо своевременно выявлять хромосомные патологии у плода. Для этого всем беременным женщинам не следует пренебрегать антенатальным скринингом.
В настоящее время в нашей стране работают специальные центры, в которых ухаживают за детьми с врожденными заболеваниями и развивают их интеллект. Если больной ребенок живет в таком центре больше года под наблюдением врачей, он начинает улыбаться, реагировать на движение, самостоятельно поддерживать положение тела и питаться.
Синдром Эдвардса – патология, которую фактически невозможно ни прогнозировать, ни лечить, ни корректировать. Данный диагноз является показанием к аборту.
Видео: о детях с синдромом Эдвардса
sindrom.info
Синдром Эдвардса — причины, симптомы, диагностика и лечение
Синдром Эдвардса — хромосомное заболевание, обусловленное трисомией по 18-ой хромосоме и сопровождающееся множественными пороками развития. Для синдрома Эдвардса характерны своеобразные фенотипические признаки (долихоцефалическая форма черепа, микрофтальмия, недоразвитие ушных раковин, микроретрогнатия и др.), аномалии опорно-двигательной, сердечно-сосудистой, пищеварительной, мочеполовой системы, ЦНС. Синдром Эдвардса может быть диагностирован на этапе беременности (УЗИ-скрининг, инвазивная пренатальная диагностика) либо уже после рождения ребенка на основании внешних признаков и цитогенетического исследования. Дети с синдромом Эдвардса нуждаются в симптоматическом лечении и хорошем уходе.
Общие сведения
Синдром Эдвардса – количественная хромосомная аберрация, при которой имеет место частичная или полная трисомия по 18 аутосоме. Синдром получил название по имени генетика J. Edwards, подробно описавшего заболевание в 1960 г. и выделившего свыше 130 характерных для данной патологии симптоматических дефектов. Синдром Эдвардса – второе по распространенности хромосомное заболевание после синдрома Дауна; частота рождения детей с синдромом Эдвардса составляет 1:5000-7000. Примерно три четверти всех больных синдромом Эдвардса – девочки; предполагается, что большая часть беременностей плодом мужского пола заканчивается внутриутробной гибелью и самопроизвольным абортом.
Синдром Эдвардса
Причины синдрома Эдвардса
Развитие синдрома Эдвардса объясняется хромосомными нарушениями, происходящими на стадии гаметогенеза (овогенеза или сперматогенеза) либо дробления зиготы и приводящими к увеличению числа хромосом 18-й пары. В 80-90% случаев цитогенетические варианты синдрома Эдвардса представлены простой трисомией 18, реже — мозаичной формой или несбалансированными перестройками (транслокациями).
Причиной полной трисомии служит мейотическое нерасхождение хромосом. Практически во всех случаях лишняя хромосома является материнской по происхождению. Этот вариант синдрома Эдвардса является наиболее тяжелым по своим проявлениям и неблагоприятным в плане прогноза. Возникновение мозаицизма связано с нерасхождением хромосом на ранней стадии дробления зиготы. В этом случае лишнюю хромосому будут содержать не все клетки плода, а лишь их часть. Транслокация – присоединение части 18-ой хромосомы к другой паре может произойти как в процессе созревания гамет, так и после оплодотворения. При этом клетки организма содержат две гомологичные 18-е хромосомы и ее дополнительную часть, прикрепленную к другой хромосоме.
Как и в случае с синдромом Дауна, возраст матери является наиболее значимым риск-фактором рождения ребенка с синдромом Эдвардса. В редких случаях у родителей может выявляться носительство сбалансированной транслокации.
Симптомы синдрома Эдвардса
Во время беременности наблюдается многоводие, слабая активность плода, маленькая плацента, единственная пупочная артерия. Ребенок с синдромом Эдвардса рождается с низкой массой тела (около 2170 г) и пренатальной гипотрофией при доношенной или даже переношенной беременности. У части детей определяется состояние асфиксии при рождении.
У новорожденных с синдромом Эдвардса имеются характерные фенотипические признаки, позволяющие предположить данную хромосомную патологию. В первую очередь обращает на себя внимание долихоцефалическая форма черепа с преобладанием продольного размера над поперечным, низкий лоб, выступающий затылок, микрогнатия, маленький рот, микрофтальмия. У детей с синдромом Эдвардса часто встречаются расщелины верхней губы и нёба, эпикант, птоз, экзофтальм, косоглазие, короткая шея с избыточной кожной складкой. Типичные деформации ушных раковин включают маленькие мочки, отсутствие козелков, узкие слуховые проходы, низкое расположение ушей.
Внешний облик детей дополняется характерными для синдрома Эдвардса деформациями скелета — скрещенными пальцами кистей, укороченной грудиной, аномалиями ребер, врожденным вывихом бедра, косолапостью, «стопой-качалкой», синдактилией стоп и пр. У многих детей имеются гемангиомы и папилломы кожи.
При синдроме Эдвардса имеются множественные тяжелые аномалии со стороны практически всех систем организма. Врожденные пороки сердца могут быть представлены дефектами межжелудочковой и межпредсердной перегородок, коарктацией аорты, транспозицией магистральных сосудов, дисплазией клапанов, тетрадой Фалло, аномальным дренажом легочных вен, декстракардией и др. При синдроме Эдвардса может выявляться патология развития желудочно-кишечного тракта: диафрагмальные, пупочные и паховые грыжи, дивертикул Меккеля, трахеопищеводные свищи, пилоростеноз, атрезия подвздошной кишки и ануса. Наиболее частыми аномалиями мочеполовой системы у детей с синдромом Эдвардса служат подковообразная почка, гидронефроз, дивертикулы мочевого пузыря, гипоспадия и крипторхизм (у мальчиков), двурогая матка, внутриматочная перегородка и гипертрофия клитора (у девочек).
Пороки развития центральной нервной системы характеризуются наличием микроцефалии, менингомиелоцеле, гидроцефалии, аномалии Арнольда-Киари, кист арахноидального сплетения, гипоплазии мозжечка и мозолистого тела. У всех выживших детей с синдромом Эдвардса имеются интеллектуальные нарушения — олигофрения в степени глубокой имбецильности или идиотии.
Новорожденные с синдромом Эдвардса испытывают трудности с сосанием, глотанием и дыханием, из-за чего им требуется зондовое питание или длительная ИВЛ. Дети с синдромом Эдвардса, как правило, погибают на первом году жизни из-за тяжелых врожденных пороков развития и связанных с ними осложнений (сердечно-сосудистой и дыхательной недостаточности, пневмонии, кишечной непроходимости и т. д.).
Диагностика синдрома Эдвардса
Важнейшей задачей диагностики служит антенатальное выявление синдрома Эдвардса у плода, поскольку данная патология является медицинским показанием для искусственного прерывания беременности. Заподозрить наличие синдрома Эдвардса можно в процессе УЗИ плода и допплерографии маточно-плацентарного кровотока по косвенным признакам (множественным аномалиям развития плода, агенезии пупочной артерии, малой величине плаценты, многоводию и пр.).
Наибольшую диагностическую значимость имеет стандартный пренатальный скрининг, включающий анализ крови на сывороточные маркеры: βХГЧ и PAPP на 11-13 неделе беременности; βХГЧ, альфа-фетопротеин и свободный эстриола на 20-24 неделе гестации.
При оценке степени риска рождения ребенка с синдромом Эдвардса учитываются данные биохимического и ультразвукового скрининга, срок беременности, возраст и масса тела женщины. Беременным, попадающим в группу высокого риска, предлагается проведение инвазивной дородовой диагностики (биопсии хориона, амниоцентеза, кордоцентеза) с последующим кариотипированием плода.
В случае рождения живого ребенка с синдромом Эдвардса необходимо как можно более раннее всестороннее обследование, направленное на выявление тяжелых пороков развития. Новорожденный с синдромом Эдвардса должен быть осмотрен неонатологом, детским кардиологом, детским неврологом, детским хирургом, детским ортопедом, детским урологом и др. Наиболее важными диагностическими исследованиями, которые должны быть выполнены ребенку с синдромом Эдвардса в первые часы жизни, служат эхокардиография, УЗИ органов брюшной полости и УЗИ почек.
Лечение синдрома Эдвардса
Поскольку в большинстве случаев аномалии развития оказываются несовместимыми с жизнью, лечение детей с синдромом Эдвардса сводится к оказанию симптоматической помощи, направленной на поддержание физиологических функций, продление жизни и улучшение ее качества. Хирургическая коррекция врожденных пороков, как правило, является рискованной и неоправданной.
Поскольку дети с синдромом Эдвардса ослаблены и подвержены частой заболеваемости инфекциями мочевыводящих путей, средним отитом, конъюнктивитом, синуситами, пневмониями и пр., они нуждаются в тщательно организованном уходе, полноценном питании, регулярном наблюдении со стороны педиатра.
Прогноз и профилактика синдрома Эдвардса
Во всех случаях прогноз при синдроме Эдвардса крайне неблагоприятный: в среднем мальчики живут 2-3 месяца, девочки – 10 месяцев. До 1 года доживает лишь 10% больных, до 10 лет – не более 1%. Относительно благоприятные шансы в отношении выживания имеют дети с мозаичной формой синдрома Эдвардса.
Риск рождения ребенка с синдром Эдвардса теоретически существует в любой супружеской паре; известно, что такая вероятность выше у возрастных родителей (для женщин старше 45 лет – 0,7%). С целью своевременного выявления хромосомной патологии у плода не следует пренебрегать антенатальным скринингом, входящим в программу введения беременности.
www.krasotaimedicina.ru
что это. Симптомы, причины, диагностика и лечение синдрома Эдвардса
Синдром Эдвардса представляет собой хромосомную патологию, которая характеризуется наличием дополнительной хромосомы (трисомия) в 18 паре.
Это тяжелая врожденная патология, вторая по частоте после синдрома Дауна, характеризующаяся тяжелыми аномалиями в развитии внутренних органов с неблагоприятным прогнозом. Согласно статистическим данным, частота болезни во всем мире находится в пределах 0,015—0,02%. На три-четыре больных девочки приходится один мальчик.
Причины синдрома Эдвардса
Синдром Эдвардса — это генетическое заболевание. Суть патологии заключается в наличии дополнительной третьей хромосомы в 18 паре.
Хромосомы — это очень компактно упакованная молекула ДНК, в которой закодированы все свойства человеческого организма. Каждая нормальная клетка содержит 46 хромосом. 44 из них являются соматическими, то есть, несут информацию о развитии и функционировании всех органов и систем, еще 2 — половые, определяющие принадлежность к тому или иному полу.
Так как синдром Эдвардса — это хромосомное заболевание, то в его основании лежит не мутация определенного гена, а дефект всей хромосомы, то есть, целой молекулы ДНК, а именно — дополнительная хромосома.
Точные причины синдрома Эдвардса не установлены. Обычно появление лишней хромосомы возможно до оплодотворения, что связано с аномалиями в генетическом материале сперматозоида или яйцеклетки. Провоцирует такое нарушение возраст родителей старше 40 лет, наличие подобных хромосомных нарушений в семье одного из родителей ребенка, а также злоупотребление алкоголем и курение одного из будущих родителей, прием лекарственных средств, ионизирующее излучение.
Вероятность развития хромосомных патологий напрямую зависит от возраста родителей, что доказано в ходе крупных научных исследований. Особенно важное значение имеет возраст матери — после 40 лет возможность зачать больного ребенка увеличивается примерно в 7 раз в сравнении с общей популяцией. Возраст отца также важен, но здесь зависимость выражена не так сильно.
Вредные вещества — этиловый спирт, сигаретный дым, наркотические соединения, при регулярном воздействии нарушают процессы созревания половых клеток и способствуют неправильному распределению хромосом. Как следствие рождается ребенок с тяжелой патологией.
Ионизирующие лучи, независимо от типа излучения, способны вызывать мутации в половых клетках. Особенно опасно влияние больших доз радиации в период полового созревания, когда в половые органы интенсивно растут и развиваются.
Узнайте экспертное мнение
Оставьте свой e-mail и мы расскажем, как правильно обследоваться и приступить к лечению
Признаки и симптомы синдрома Эдвардса
Симптомы синдрома Эдвардса во многом зависят от формы заболевания у конкретного больного.
На сегодняшний день генетики различают три типа болезни:
- полная трисомия;
- частичная трисомия;
- мозаичный синдром Эдвардса.
Полная трисомия — наиболее тяжелая форма, при которой симптомы синдрома Эдвардса выражены сильнее всего. В этом случае дополнительная хромосома содержится во всех клетках организма больного. Этой формой страдает до 90% больных.
Частичная трисомия диагностируется только у 3 больных из 100. В этой ситуации клетки содержат лишь фрагмент восемнадцатой хромосомы. Заболевания имеет место, если причиной патологии стали нарушения в процессе размножения половых клеток и, как следствие, неправильное распределение ДНК. Прогноз при частичной трисомии несколько лучше, чем при полной.
На мозаичную форму приходится до 7% случаев. В этом случае болезнь развивается после зачатия, при условии, что на плодное яйцо воздействовали неблагоприятные факторы. Именно в этом случае причины синдрома Эдвардса — химические вещества и физические факторы, поступающие извне. Как известно, уже в первые минуты после оплодотворения в яйцеклетке начинаются интенсивные процессы деления. Если в них произойдет сбой, то при очередном делении клетка получит дополнительную хромосому. В дальнейшем, она станет источником таких же дефектных клеточных элементов. Максимальное количество аномальных клеток при мозаичной форме синдрома Эдвардса — 50%.
Дети в случае наличия такого изменения количества хромосом рождаются со сниженной массой тела и наличием множественных пороков развития, которые заключаются в изменении формы черепа, уменьшении диаметра рта, аномалии твердого неба, наличии узких глазных щелей, деформации ушных раковин, дефекты конечностей, половых органов, специфические дерматоглифические проявления. Также часто развиваются пороки сердца, которые заключаются в нарушении заращения перегородок и формирования клапанов между его полостями. Благодаря яркой клинической картине диагностика заболевания не представляет серьезных трудностей.

Долихоцефалия — типичный признак заболевания. Череп больных детей имеет узкую и удлиненную форму, что подтверждают специальные измерения (соотношение ширины черепа к его длине). В норме показатель должен быть не менее 75%. Иногда эта форма встречается и у здоровых малышей, но если изменение заметно невооруженным глазом, без измерений, то вероятность того, что ребенку будет поставлен диагноз «синдром Эдвардса», очень велика.
Еще один внешний признак патологии — микроцефалия, при котором голова очень мала по отношению к туловищу. Особенно сильно недоразвита мозговая часть черепа.
У 95% больных детей наблюдается аномалия ушной раковины. Хрящ, который ее формирует, плоский. Выпуклости, характерные для здоровых людей, отсутствуют или выражены очень слабо, а сами раковины расположены низко. Нередко отсутствуют мочки уха или козелок. Слуховой ход сужен или атрезирован. Полное отсутствие слухового хода наблюдается у каждого 4 больного.
Еще одна характерная черта патологии — волчья пасть. Это врожденная аномалия развития твердого неба, которая появляется из-за несрастания небных отростков верхней челюсти.
В зависимости от степени выраженности различают 4 варианта порока:
- несрастание только мягкого неба;
- несрастание мягкого и части твердого неба;
- полное несрастание твердого неба;
- полное несрастание мягкого, твердого неба и губы.
Деформация стопы в виде стопы-качалки обусловлена неправильным расположением костей. При этом пятка сильно выступает назад, свод стопы отсутствует. Вогнутая линия, которая в норме соединяет пятку и большой палец, отсутствует, нередко стопа выпуклая, из-за чего похожа на ножки кресла-качалки, что и обусловило название деформации.
При наличии стопы-качалки часто наблюдается непропорциональное строение пальцев ног. Большой палец у больных детей короткий — короче, чем второй палец стопы. Важно понимать, что у взрослых этот признак не имеет диагностической ценности, так как может появиться из-за других заболеваний, неправильной обуви, суставной аномалии. Вместе с непропорциональным строением нередко имеет место сращение пальцев. В легких случаях это лишь кожная складка, но нередко обнаруживают и костные дефекты.
Кисти малыша находятся в специфическом положении — они сжаты в кулак, но мизинец и указательный палец при этом располагаются поверх среднего и безымянного. Это так называемое флексорное положение кисти, обусловленное повышенным тонусом мышц-сгибателей.
Недоразвитая нижняя челюсть встречается у 70% детей, которым поставлен диагноз «синдром Эдвардса». Подбородок сильно втянут, что заметно невооруженным глазом. Ребенок не может держать рот закрытым, что ведет к вытеканию слюны. У выживших детей формируется неправильный прикус.
Из аномалий половых органов чаще всего имеют место недоразвитие полового члена и увеличение клитора. Также возможно неправильное расположение уретры. У мальчиков яички могут оставаться в брюшной полости или паховом канале. В большинстве случаев эти отклонения сочетаются с пороками внутренних половых органов и мочевого пузыря.
Дерматоглифические симптомы — характерные отклонения в расположении узоров и складок на ладонях. В большинстве случаев дерматоглифика используется как предварительная диагностика синдрома Эдвардса, особенно частичной и мозаичной форм. При полной трисомии клиническая картина выражена настолько ярко, что потребность в детальном изучении узора ладоней отпадает за ненадобностью.
Важно понимать, что сами по себе вышеперечисленные признаки — далеко не диагноз. Единичные отклонения могут быть вариантом нормы у абсолютно здоровых людей, но наличие нескольких наиболее часто встречающихся отклонений является поводом для более детального обследования.
Диагностика синдрома Эдвардса
Диагностика синдрома Эдвардса заключается в генетическом исследовании, которое может проводиться во время беременности. При этом выполняется цитологическое исследование околоплодных вод с определением количества хромосом клеток эпидермиса плода. Так как подобные манипуляции провоцируют определенные осложнения, синдром Эдвардса с их помощью диагностируют только пациентам из группы риска.
Лечение синдрома Эдвардса
На данный момент лечение синдрома Эдвардса невозможно, так как хромосомная аномалия затрагивает все клетки плода или родившегося ребенка. Поэтому важно диагностировать синдром Эдвардса на ранних сроках течения беременности.
В случае подтверждения диагноза возможно проведение прерывания беременности по медицинским показаниям. Для родившегося ребенка с таким хромосомным нарушением прогноз неблагоприятный. Большинство детей не доживают до годовалого возраста. Выжившие дети с синдромом Эдвардса значительно отстают в развитии (особенно в умственном) и требуют постоянного медицинского наблюдения. В таком случае на первый план выходит тщательный уход и симптоматическое лечение синдрома Эдвардса.
Запишитесь на приём прямо сейчас
Профилактика синдрома Эдвардса
Профилактика синдрома Эдвардса сводится к тщательному планированию предстоящей беременности. Всем парам из группы риска настоятельно рекомендуют проконсультироваться у генетика. Врач детально изучит семейный анамнез пары, выявит факторы риска, проведет генетический анализ пары.
При сборе семейного анамнеза доктор расспросит о наследственных заболеваниях в семье. При наличии таковых, вероятность рождения ребенка с аномалией возрастает до 1%. Очень желательно, чтобы родители собрали максимум информации о своих предках.
Затем генетик выявит факторы риска и даст рекомендации по их устранению, если это возможно.
Генетический анализ необходим для обнаружения дефектных генов в молекуле ДНК. Чем больше мутаций будет выявлено, тем выше вероятность рождения больного ребенка.
Всем беременным из группы риска показана пренатальная диагностика, благодаря которой можно вовремя диагностировать болезнь и принять решение по поводу дальнейшего вынашивания ребенка. В принципе, это также профилактика синдрома Эдвардса.
Из-за множественных пороков развития примерно половина детей умирает в первые три месяца жизни. Только 10 детей из 100 доживут до года. Более старшие дети нуждаются в тщательном уходе, постоянном лечении и профилактике осложнений синдрома Эдвардса. При мозаичной или частичной форме прогноз более благоприятный. Длительность жизни детей больше, но они также нуждаются в уходе. При полноценных реабилитационных мероприятиях малыш может научиться сидеть и питаться, иногда хорошо выражена реакция на окружающих. Из-за серьезной умственной отсталости больные не способны устроиться даже на простейшую работу.
Синдром Эдвардса можно предотвратить, если вы беременеете с помощью ЭКО. В клинике «АльтраВита» практикуется диагностическая процедура под названием ПГД (преимплантационная генетическая диагностика). Она дает возможность выявить хромосомную аномалию еще до помещения эмбриона в полость матки, то есть, до наступления беременности. Для переноса будет выбран тот эмбрион, который гарантированно не имеет никаких генных или хромосомных мутаций. Позвоните в клинику «АльтраВита», чтобы узнать о возможности проведения ПГД.
altravita-ivf.ru
Синдром Эдвардса: признаки, причины и диагностика
Внешние и внутренние проявления заболевания могут существенно различаться в зависимости от особенностей развития плода. Чаще всего хромосомные нарушения появляются на начальных этапах развития эмбриона, поэтому они сказываются на развитии всего организма. Есть несколько внешних признаков, по которым с большой вероятностью можно предположить наличие синдрома Эдвардса у новорожденных.
Одной из наиболее характерных черт этого заболевания является искаженная форма черепных костей: череп вытянут от макушки к подбородку, вместе с тем часто ставится диагноз «микроцефалия» (уменьшение размеров черепа и мозга) или «гидроцефалия» (скопление жидкости в головном мозге). Лоб узкий, а затылочная сторона более широкая и выступающая, при этом уши расположены ниже, чем при нормальном развитии. Деформируются челюстные кости – часто это приводит к значительному уменьшению нижней челюсти, она становится узкой и недоразвитой. Вследствие этого рот также небольшой и часто треугольный из-за укорочения верхней губы. Небо высокое, иногда присутствует щель. Шея может быть укороченной, с характерной складкой.
Глазные щели уже и короче, чем нужно, переносица расширена и вдавлена – это особенно заметно при том, что нос, как правило, заужен, косточки носа могут визуально отсутствовать. Глазное яблоко также подвержено изменениям и нарушениям, которые приводят к катаракте и колобоме, то есть отсутствию части глазной оболочки. Кроме того, могут быть и иные нарушения зрения.
Уши низко посажены и деформированы, часто в горизонтальной плоскости. На ухе часто отсутствует мочка, а иногда козелок. Наружный слуховой проход часто сужен, иногда может отсутствовать полностью.
Широкий спектр нарушений касается костного скелета. Прежде всего, суставы не могут функционировать нормально, поэтому стопы и кисти не могут сгибаться и разгибаться так, как нужно. К тому же стопы недоразвиты, из-за этого изменяется их форма, они становятся менее подвижными. Большой палец укорочен, а второй и третий срастаются, иногда настолько сильно, иногда формируются ластообразные конечности. В 80% случаев формируется стопа с провисающим сводом, выступающей пяткой и коротким большим пальцем.
Из-за излишней подвижности тазобедренного сустава нередко случаются вывихи.
На подушечках пальцев количество дуг может быть в 10 раз больше нормы, однако сгибательной складки пальцы не имеют. Почти у 30% больных на ладонях появляются поперечные борозды и множество гребешков.
Помимо всего прочего, при синдроме Эдвардса деформируется форма грудной клетки – она расширяется, а межреберные промежутки уменьшаются, таким образом она становится короче и шире.
Значительные изменения претерпевают и внутренние органы. Почти все пациенты имеют порок сердца. Как правило, он характеризуется недостаточной развитостью клапанов в артериях и аортах. При этом довольно часто появляется дефект межжелудочковой перегородки.
Имеют место очень серьезные нарушения в обменных процессах, таких как работа эндокринной системы. Вследствие хромосомных нарушений железы не могут функционировать нормально, поэтому рост существенно замедляется. Гормональные нарушения приводят к недоразвитости подкожной клетчатки. У каждого десятого отмечается нарушение работы надпочечников или щитовидной железы.
Пониженный мышечный тонус со временем обычно повышается, при этом улучшается кровообращение.
Примерно у половины больных наблюдается аномальное развитие кишечника. Чаще всего эта аномалия заключается в его необычном расположении, при этом появляется мешок, образованный из слоев стенки кишки, а пищевод сужается слишком резко. Почки часто бывают сегментированы или имеют неправильную форму дуги, также может быть удвоение мочеточников.
Изменения также затрагивают и половые органы. У мальчиков яичко может не опускаться в мошонку (крипторхизм) и меняется строение пениса. У девочек формируется гипертрофированный клитор, а яичники развиты недостаточно.
В целом картина внешних и внутренних отклонений при синдроме Эдвардса выглядит следующим образом. В 100% случаев наблюдаются аномалии строения черепа и изменение формы лица. Почти у 97% уменьшение челюсти (микрогения), чуть более чем в 95 % случаев нарушается строение и расположение ушных раковин. Удлинение черепа наблюдается почти у 90% пациентов, высокое небо – у 78%, а уменьшенный рот – в 71% случаев.
Что касается нарушений конечностей, то они есть у 98% больных. Чаще всего встречается изменение формы кистей (более 91%) и стоп (76%).
Развитие сердечно-сосудистой системы нарушено более чем у 90% пациентов. Около 1/3 пациентов имеют нарушения мочеполовой системы, а 55% –пищеварительной системы.
panoramatest.ru
Синдром Эдвардса — причины, риски, признаки на УЗИ
Большинство женщин хорошо знают, что такое синдром Дауна. И во время беременности многие узнают, что очень редко, но встречается и другое хромосомное нарушение, называемое Синдром Эдвардса. И многие обеспокоены, как узнать насколько высок риск рождения ребенка с синдромом Эдвардса и как диагностируется такая патология при беременности?
Что такое Синдром Эдвардса?
Синдром Эдвардса — это генетическое заболевание, характеризующееся дублированием (трисомией) ХVIII хромосомы и проявляющееся целым рядом характерных пороков развития у плода во время беременности, часто приводящих к смерти ребенка или его инвалидизации. То есть у ребенка вместо 46 хромосом образуется 47, эта лишняя хромосома дает другое название болезни — трисомия 18. Синдром был назван в честь исследователя Джона Эдвардса, который впервые описал его в 1960 году.
Почему возникает синдром Эдвардса — причины патологии
Даже если родители здоровы, и в семейном анамнезе нет такой патологии, ребенок с 18 хромосомой может родиться у любой женщины.
Как известно, в каждой человеческой клетке находится 46 хромосом, а в женских и мужских половых клетках по 23 хромосомы, которые, соединяясь при оплодотворении яйцеклетки, также дают в сумме 46 хромосом. Если говорить о синдроме Эдвардса, причины его появления неизвестны.
В настоящее время известно только то, что в результате определенных генетических мутаций появляется лишняя 47–я хромосома (дополнительная хромосома в 18–й паре хромосом, которых таким образом становится не 2, а 3). 
В 95% всех случаев развития синдрома Эдвардса в клетках находится именно лишняя 18–я хромосома (трисомия), однако в 2% наблюдается только «удлинение» 18–й хромосомы (транслокация), когда общее число хромосом остается нормальным и равняется 46.
В 3% случаев синдрома Эдвардса имеет место «мозаичная трисомия», когда дополнительная 47–я хромосома обнаруживается в организме не во всех клетках, а лишь определенной его части. Клинически все 3 варианта синдрома Эдвардса протекают практически одинаково, однако первый вариант может отличаться более тяжелым течением заболевания.
Как часто встречается эта патология?
Дети с синдромом Эдвардса погибают на этапе внутриутробного развития приблизительно в 60% случаев. Несмотря на это, среди генетических заболеваний данный синдром у выживших младенцев является достаточно распространенным, по частоте встречаемости уступая только синдрому Дауна. Распространенность синдрома Эдвардса составляет 1 случай на 3 — 8 тысяч детей.
Считается, что у девочек данное заболевание встречается в 3 раза чаще, а риск синдрома Эдвардса значительно возрастает, если беременной женщине 30 и более лет.
Смертность при синдроме Эдвардса на первом году жизни составляет около 90%, причем средняя продолжительность жизни при тяжелом течении заболевания у мальчиков — 2–3 месяца, а девочек — 10 месяцев, а до взрослого состояния доживают лишь единицы. Чаще всего дети с синдромом Эдвардса умирают от удушья, пневмонии, сердечно–сосудистой недостаточности или кишечной непроходимости — осложнений, вызванных врожденными пороками развития.
Как проявляется синдром у ребенка?
Признаки синдрома Эдвардса можно разделить на несколько больших групп:
К первой группе относятся симптомы, характерные для внешнего вида ребенка:
- При рождении низкая масса тела (около 2 100 – 2200 граммов)
- Непропорционально маленькая голова
- Дефекты развития верхней или нижней челюстей (микрогнатия)
- Искажение формы лица и формирование неправильного прикуса
- «Волчья пасть» (расщелина твердого неба) или «заячья губа» (расщелина верхней губы)
- Пальцы кисти сжаты, в кулаке располагаются неровно
- Низкая посадка ушей
- Перепончатость или полное слияние пальцев нижних конечностей
- Врожденная косолапость
- «Стопа–качалка»
- Относительно малых размеров ротовая щель (микростомия)
Ко второй группе можно отнести признаки нарушения работы внутренних органов, моторики и нервно–психического развития:
- Наличие врожденных пороков сердца, например, открытого овального отверстия, дефекта межжелудочковой перегородки, открытого артериального протока и т.п.
- Развитие паховых или пупочных грыж.
- Органы пищеварения: гастроэозофагиальная рефлюксная болезнь, нарушение глотательного и сосательного рефлекса, атрезия пищевода или заднего прохода, меккелев дивертикул, нарушение расположения кишечника.
- Центральная нервная система: задержка нервно–психического развития, умственная отсталость, недоразвитость мозжечка, мозолистого тела, сглаживание или атрофия мозговых извилин.
- Мочеполовая система: крипторхизм, гипоспадия у мальчиков, гипертрофия клитора, недоразвитость яичников у девочек, независимо от пола — подковообразная или сегментированная почка, удвоение мочеточников.
- Косоглазие, сколиоз, атрофия мышц.
Как узнать патологию во время беременности — диагностика
Синдром Патау, Эдвардса и другие трисомии лучше всего выявить до рождения малыша. Как правило, пренатальная диагностика данного синдрома проводится в 2 этапа:
- На сроке в 11–13 недель (скрининг, в основе которого — проведение различных биохимических анализов у женщины).
- Определение кариотипа плода у беременных их группы риска.
В 11–13 недель в крови женщины определяется уровень некоторых белков крови: β–ХГЧ (β–субъединица хорионического гормона человека) и плазменного протеина А ассоциированного с беременностью. Затем с учетом этих данных, возраста беременной женщины рассчитывается риск рождения ребенка с синдромом Эдвардса, и формируется группа риска беременных.
Далее в группе риска на более позднем сроке берется материал у плода для постановки точного диагноза: в 8–12 недель это биопсия ворсин хориона, в 14–18 — амниоцентез (изучение околоплодных вод), спустя 20 недель — кордоцентез (внутриутробное взятие крови из пуповины плода с УЗИ–контролем). После этого в полученном материале определяют наличие или отсутствие дополнительной 18–й хромосомы с помощью КФ–ПЦР (количественной флуоресцентной полимеразной цепной реакции).
Если беременная не прошла генетическое скрининг–обследование, то на более поздних сроках предварительная диагностика синдрома Эдвардса осуществляется с помощью УЗИ. Прочие косвенные признаки, на основании которых можно заподозрить синдром Эдвардса на более поздних сроках:
- Наличие аномалий развития костей и мягких тканей головы («волчья пасть», микроцефалия, низкая посадка ушных раковин, «заячья губа» и т.п.).
- Обнаружение пороков со стороны сердечно–сосудистой , мочеполовой системы, а также опорно–двигательного аппарата.
Диагностические признаки синдрома у ребенка
После рождения ребенка опорными диагностическими признаками наличия синдрома Эдвардса являются следующие:
- Микроцефалия, малый вес при рождении
- Наличие «заячьей губы» или «волчьей пасти»
Признаки характерной дерматографической картины:
- неразвитая на пальцах дистальная сгибательная складка
- наличие в 1/3 случаев поперечной ладонной борозды
- дуги на подушечках пальцев рук
- изменение кожного рисунка ладони: дистальное расположение осевого трирадиуса и увеличение гребневого счета.
Далее диагноз также подтверждается с помощью определения кариотипа ребенка методом КФ–ПЦР.
Синдром Эдвардса на УЗИ — кисты сосудистых сплетений
На ранних сроках синдром Эдвардса на УЗИ заподозрить крайне трудно, однако в 12 недель беременности уже выявляют характерные для него симптомы косвенного характера:
- Признаки задержки развития плода
- Брадикардия (снижение у плода частоты сердечных сокращений)
- Омфалоцеле (наличие грыжи брюшной полости)
- Отсутствие визуализации косточек носа
- В пуповине одна, а не 2 артерии
Также на УЗИ могут быть обнаружены кисты сосудистых сплетений, представляющих собой полости с содержащейся в них жидкостью. Сами по себе они не несут угрозу для здоровья и исчезают к 26–недельному сроку беременности. Однако такие кисты очень часто сопровождают различные генетические заболевания, например, синдром Эдвардса (в данном случае кисты обнаруживаются у 1/3 детей, страдающих этой патологией), поэтому при обнаружении таких кист врач направит беременную женщину на обследование в генетическую консультацию.
Лечение
Так как дети с синдромом Эдвардса редко доживают до года, то сначала лечение направлено на коррекцию тех пороков развития, которые опасны для жизни:
- восстановление прохода пищи при атрезии кишечника или анального отверстия
- кормление через зонд в случае отсутствия глотательного и сосательного рефлексов
- антибактериальная и противовоспалительная терапия при воспалении легких
При относительно благоприятном течении заболевания происходит коррекция некоторых аномалий и пороков развития: хирургическое лечение «волчьей пасти», пороков сердца, паховой или пупочной грыжи, а также симптоматическое медикаментозное лечение (назначение слабительных при запорах, «пеногасителей» при метеоризме и т.п.). 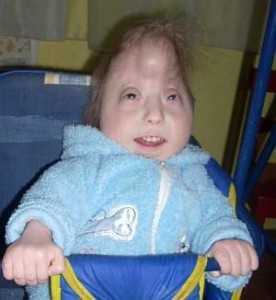
Дети с синдромом Эдвардса склонны к таким заболеваниям, как:
- средний отит
- рак почки (опухоль Вильмса)
- пневмония
- конъюнктивит
- легочная гипертензия
- апноэ
- повышенное артериальное давление
- фронтиты, синуситы
- инфекции мочеполовой системы
Поэтому лечение больных с синдромом Эдвардса включает своевременную диагностику и терапию данных заболеваний.
Прогноз для ребенка
В большинстве случаев прогноз неблагоприятный. Единицы детей с синдромом Эдвардса, которые доживают до взрослого возраста, имеют серьезные умственные отклонения и постоянно требуют постороннего ухода и контроля. Однако при соответствующих занятиях они способны реагировать на утешение, улыбаться, самостоятельно принимать пищу, а также взаимодействовать с воспитателями, приобретая различные умения и навыки.
zdravotvet.ru
Трисомия 18. Симптомы, диагностика, лечение
Трисомия 18 была независимо описана Эдвардсом и доктором Смиттом в 1960. Среди живорожденных детей, трисомия 18 является второй наиболее распространенной трисомией после трисомии 21. Это расстройство характеризуется развитием тяжелой психомоторной задержки роста, микроцефалией, микрофтальмией, аномальными ушами, микрогнатией или ретрогнатией, отчетливо сжатыми пальцами и другими врожденными пороками развития.
Трисомия 18. Причины
- Приблизительно в 90% случаев, дополнительная хромосома имеет материнское происхождение (ошибки на первой фазе мейоза). Это очень характерное отличие трисомии 18 от других человеческих трисомий, которые проявляют более высокую частоту в первой фазе мейоза. Полная трисомия 18 встречается у 95% детей с этим типом трисомии.
- В оставшихся случаях встречается мозаичная и транслокационная трисомия 18. Клинический фенотип, при мозаичной трисомии 18, может очень сильно изменяться, в зависимости от уровня мозаичности и тканей, в клетках которых наблюдается аномальное количество хромосом.
- Транслокационная трисомия приводит к частичной трисомии 18. Частичная трисомия 18 появляется тогда, когда сегмент хромосомы 18 будет присутствовать в трех, а не в двух экземплярах. Это часто происходит в результате сбалансированной транслокации у одного из родителей. На этот тип трисомии 18 приходится примерно 2% от всех трисомий 18.
Трисомия 18. Патофизиология
Трисомии 18 серьезно влияют на все органы и системы. Транслокационные и мозаичные трисомии 18 харрактеризуются развитием менее тяжелых клинических проявлений и прогноз для пациента, как правило, будет более благоприятным.
Трисомия 18. Симптомы и проявления
Неврологические проявления
- Задержка психомоторного развития и умственная отсталость у 100% пациентов
- Неонатальная гипотония сопровождающаяся гипертонией, апноэ, судорогами
- Пороки развития (например, микроцефалия, гипоплазия мозжечка, анэнцефалии, гидроцефалия, гипоплазия или аплазия мозолистого тела, аномалии серпа мозга, дефекты лобных долей мозга, арахноидальные кисты, миеломенингоцеле)
Череп – микроцефалия, удлиненный череп, широкие роднички и видный затылок
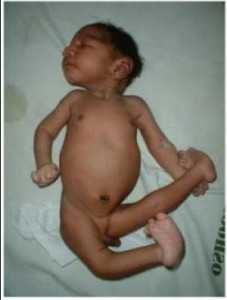
Фото ребенка с характерными особенностями трисомии 18 — сжатые перекрывающиеся пальцы, аномалии нижних ног, микрогнатия, низко посаженные уши
Лицо – микрофтальмия, глазной гипертелоризм, короткие глазные щели, колобомы, катаракты, помутнение роговицы, нарушения в пигментации сетчатки, короткий нос с приподнятыми ноздрями, атрезии хоан, микрогнатия или ретрогнатия, узкие небные дуги, заячья губа, волчье нёбо, аномальные уши.
Скелет – тяжелая задержка роста, характерные позы (очень часто встречаются такие ситуации, при которых некоторые пальцы будут перекрывать собой соседние, например указательный палец может перекрывать средний, а пятый палец будет перекрывать четвертый палец), радиальные гипоплазии или аплазии, синдактилии второго и третьего пальцев, артрогрипоз, косолапость, гипоплазии ногтей, короткая шея с чрезмерным количеством складок кожи, короткая грудина, узкий таз.
Сердце
- Более 90% детей с трисомией 18 имеют сердечные пороки.
- Наиболее распространенные аномалии включают дефекты межжелудочковой перегородки с дефектами клапанов.
- Другие пороки сердца включают дефекты межпредсердной перегородки, открытый артериальный проток, коарктации аорты, тетрада Фалло и транспозиции магистральных артерий.
Легкие — гипоплазия легких
ЖКТ — пупочная грыжа, мальротация кишечника, атрезия подвздошной кишки, дивертикул Меккеля, атрезия пищевода с или без трахеопищеводного свища, диафрагмальная эвентрация, диастаз прямых мышц живота, отсутствие желчного пузыря, аномалии в селезенке, экстрофии клоаки, стеноз привратника, заращение или неправильное позиционированные ануса, грыжи (т.е., пупочная, паховая, диафрагмальная)
Мочеполовая система
- Аномалии почек, мочеточников, гидронефроз, подковообразные почки и односторонние почечные агенезии
- Крипторхизм, гипоспадия и микропенис у пациентов мужского пола
- Гипоплазия половых губ и яичников, матки, гипертрофии яичников и клитора у женщин
Эндокринная система – гипоплазия тимуса, гипоплазия щитовидной железы, надпочечников
Кожа – поперечные ладонные складки, клинодактилия пятых пальцев
Фенотипический спектр мозаичной трисомии 18:
- Фенотип лиц с мозаичной трисомией 18, в целом, широко варьируется. Некоторые люди, которые имеют полную трисомию 18 (синдром Эдвардса), как правило умирают в ранние годы жизни, тогда как другие, фенотипически вполне нормальные. Бывали даже случаи, когда мозаичная трисомия 18 обнаруживалась уже у взрослых людей, которые не проявляли никаких внешних признаков хромосомных аномалий.
- Но вышеописанный пример является скорее исключением, чем правилом. Большинство же пациентов с мозаичной трисомией 18 имеют широкий спектр аномалий, но не таких тяжелых, как те которые в встречаются у лиц с полной трисомией. К таким аномалиям можно отнести микроцефалию, брахидактилии, врожденные пороки сердца, задержки развития, низкорослость и преждевременные недостаточности яичников.
- Интеллектуальные возможности варьируются от глубокой интеллектуальной инвалидности до интеллекта выше среднего.
Трисомия 18. Диагностика
Неинвазивный скрининг в первый триместр, на основе возраста матери, наличия маркеров в сыворотке крови и сонографических «мягких маркеров», продемонстрировали высокую чувствительность в диагностике трисомии 18. Низкие уровни человеческого хорионического гонадотропина (ХГЧ) и низкие уровни неконъюгированного эстриола (UE3) в сыворотке крови матери также являются полезными предикторами повышенного риска трисомии 18.
Амниоцентез рекомендуется проводить при беременности в 14-16 недель и когда будет подозреваться трисомия 18. Точность этой процедуры составляет 99,5%. Риск прерывания беременности после проведения процедуры — небольшой (около 1 на 200-300 случаев).
Биопсия хориона выполняется на сроке в 10-13 недель. Точность составляет 96-98%.
Пренатальная диагностика трисомии 18, с помощью FISH-метода, проводится редко. Это связанно с ошибками возникающими при гибридизации α-сателлитной ДНК.
УЗИ
Большинство плодов с трисомией 18 имеют обнаруживаемые структурные аномалии. Ультрасонографические нарушения включают микроцефалию и мальформации Денди-Уокера. На УЗИ также можно обнаружит аномалии ЖКТ. У плода, как правило, можно обнаружить перекрывающиеся пальцы и косолапость. Частоты врожденных аномалий обнаруживаемых с помощью УЗИ, таковы:
- Стойкое ненормальное положение пальцев плода — 89%
- Кисты сосудистого сплетения — 43%
- Аномальные формы головы плода (форма напоминает клубнику или лимон) – 43%
- Две пуповины – 40%
- Сердечные дефекты — 37%
- Задержки внутриутробного роста – 29%
- Пупочная грыжа – 20%
- Дефекты нервной трубки – 9%
- Кистозные гигромы или лимфангиэктазии — 14%
- Многоводие — 12%
- Почечные дефекты – 9%
Эхокардиография плода
Аномальные результаты эхокардиографии обнаруживаются у большинства пациентов с трисомией 18. Сердечные пороки можно надежно диагностировать даже в первом триместре, на момент измерения затылочной полупрозрачности.
Послеродовая диагностика
Гематологические нарушения у больных с трисомией 18, в течение первой недели жизни, включают:
- Тромбоцитопения: это наиболее распространенная гематологическая аномалия, которая происходит у 83% пациентов с трисомией 18, некоторые пациенты нуждаются в переливании тромбоцитов.
- Нейтропения: это вторая наиболее часто обнаруживаемая аномалия.
- Аномальные значения эритроцитов: это третья наиболее распространенная гематологическая аномалия. Только около 45% пациентов, с трисомией 18, имеют нормальные значения эритроцитов. Анемия была обнаружена у 40%, полицитемия была обнаружена у 17%.
Трисомия 18. Лечение
Терапии трисомии 18 не существует, все усилия должны быть брошены на поддержание состояния пациента, если на это будет желание семьи. По крайней мере, 5-10% из всех пациентов выживает и очень часто (в пределах этих 5-10%), родители, в долгосрочной перспективе отказываются от своих детей. В общем, первые вмешательства должны быть сосредоточены на устранение аномалий сердечно-сосудистой системы. Инфекции необходимо лечить соответствующими препаратами (чаще всего встречается средний отит, инфекции верхних дыхательных путей, например, бронхит, пневмония и инфекции мочевыводящих путей). Сепсис необходимо лечить агрессивными терапиями, так как он имеет высокую частоту смертности. Назогастральное и гастростомическое кормление можно применять при проблемах с ЖКТ. Большинство из всех детей с трисомией 18 нуждаются в постоянном приеме мочегонных препаратов и дигоксина при застойной сердечной недостаточности.
Лечение проблем с сердцем включает в себя:
- Интенсивное фармакологическое вмешательство, паллиативная и корректирующая кардиохирургии могут улучшить выживаемость у пациентов с трисомией 18.
- В одном из исследований пациентов с трисомией 18, которые имели проблемы с сердцем, 82% пациентов, перенесших операцию на сердце были выписаны домой с облегчением сердечных симптомов. Один пациент с врожденными пороками сердца умер.
Хирургическое вмешательство
Из-за крайне плохого прогноза, хирургическое вмешательство в тяжелые врожденные аномалии, в такие как атрезии пищевода или врожденные пороки сердца, должны быть обсуждены с семьями.
Психосоциальная помощь
- В неонатальном периоде, вопросы диагностики и выживания имеют первостепенное значение. Родители всегда нуждаются в информации о синдроме, в том числе о его причинах развития, последствиях и о возможных исходах.
- Во многих западных странах, таким родителям предлагают пройти комплексный скорбный процесс, который сочетает в себе как реакреацию от горя, которая преобладает при хронической болезни, так и моральную подготовку, связанную с предстоящей смертью ребенка.
Трисомия 18. Осложнения
- Инфекции.
- Сколиоз.
- Проблемы с кормлением.
- Врожденные пороки сердца с застойной сердечной недостаточностью является частой причиной смерти.
- Опухоль Вильмса и гепатобластомы могут развиваться у некоторых лиц с трисомией 18 и у некоторых пациентов с дупликацией 18q.
Трисомия 18. Прогноз
Только небольшое количество детей с трисомией 18 доживает до своего первого года рождения и лишь немногие доживают до подросткового возраста.
- Новорожденные имеют 40% шанс дожить до 1 месяца.
- Младенцы имеют 5% шанс дожить до 1 года.
- Дети имеют 1% шанс дожить до 10 лет.
Высокая смертность развивается из-за врожденных пороков сердца, аномалий ЖКТ и мочеполовой системы, из-за трудностей в кормлении и из-за дефектов ЦНС. Хотя все дети функционируют с серьезными недостатками, все старшие дети с трисомией 18 могут улыбаться, смеяться, взаимодействовать с людьми и некоторые даже могут добиться некоторого улучшения в психомоторных отклонениях. С мозаичной формой люди имеют не такие сильные аномалии и они, как правило, могут прожить до возраста >25 лет.
redkie-bolezni.com