
Буллезного эпидермолиза: Буллезный эпидермолиз: виды, диагностика, лечение :: Здоровье :: РБК Стиль
ВРОЖДЕННЫЙ БУЛЛЕЗНЫЙ ЭПИДЕРМОЛИЗ У ДЕТЕЙ: ВОПРОСЫ ЭТИОПАТОГЕНЕЗА, КЛИНИКИ, ДИАГНОСТИКИ И ЛЕЧЕНИЯ | Сомова
1. Кубанов А. А., Альбанова В. И., Карамова А. Э. и др. Распространенность врожденного буллезного эпидермолиза у населения Российской Федерации // Вестн. дерматологии и венерологии. 2015. № 3. С. 21–30.
2. The Epidermolysis Hereditary Bullous // Orphanet Consumer
3. Encyclopedia – First edition: Sept 2012. URL: www.orpha.net/data/patho/Pub/f /EpidermolyseBulleuseHereditaire-FRfrPub11387.pdf (дата обращения: 13.06.2020).
4. Снарская Е. С., Кряжева С. С., Карташова М. Г., Бобров М. А., Филатова И. В. Врожденный дистрофический гиперпластический буллезный эпидермолиз Коккейна – Турена // Рос.
5. Fine J. D., Bruckner-Tuderman L., Eady R. A. et al. Inherited
6. Epidermolysis Bullosa: Updated Recommendations on Diagnosis
7. and Classification // J Am Acad Dermatol. 2014. № 6. Р. 1103–1126.
8. Коталевская Ю. Ю., Марычева Н. М. Буллезный эпидермолиз: основные клинические проявления // Педиатрия. № 4. 2014. С. 70–72.
10. Fine J. Inherited Epidermolysis Bullosa // Orphanet J Rare Dis. 2010. № 5. P. 12. DOI 10.1186/1750-1172-5-12.
Fine J. Inherited Epidermolysis Bullosa // Orphanet J Rare Dis. 2010. № 5. P. 12. DOI 10.1186/1750-1172-5-12.
11. Fine J. D. Epidermolysis Bullosa Registries and the Epidemiology of Epidermolysis Bullosa (EB) // Blistering Diseases. 2015. № 1. P. 265–274. DOI 10.1007/978-3-662-45698-9_22.
12. Новиков П. В. Правовые аспекты редких (орфанных) заболеваний в России и в мире // Медицина. 2013. № 4. С. 50–70.
13. Федеральные клинические рекомендации по ведению больных врожденным буллезным эпидермолизом. М., 2015. 24 с.
14. Родин А. Ю., Сердюкова Е. А., Щава С. Н. Неинфекционные буллезные дерматозы. Волгоград : Изд-во ВолгГМУ, 2013. 132 с.
 Kiritsi D., Pigors M., Tantcheva-Poor I., Wessel C., Arin M. J., Kohlhase J. et al. Epidermolysis Bullosa Simplex Ogna Revisited // J Invest Dermatol. 2013. № 133. P. 270–273.
Kiritsi D., Pigors M., Tantcheva-Poor I., Wessel C., Arin M. J., Kohlhase J. et al. Epidermolysis Bullosa Simplex Ogna Revisited // J Invest Dermatol. 2013. № 133. P. 270–273.16. Yuen W. Y., Pasmooij A., Stellingsma C., Jonkman M. F. Enamel Defects in Carriers of a Novel LAMA3 Mutation Underlying Epidermolysis Bullosa // Acta Derm Venereol. 2012. № 92. Р. 695–696.
17. Fine J. D., Bruckner-Tuderman L., Eady R. A. J. et al. Inherited Epidermolysis Bullosa: Updated Recommendations on Diagnosis and Classification // J Am Acad Dermatol. 2014. Vol. 70. № 6. P. 1103–1126.
18. Wilson N. J., Messenger A. G., Leachman S. A., O’Toole E. A., Lane E. B., McLean W. H., Smith F. J. Keratin K6c Mutations Cause Focal Palmoplantar Keratoderma // J Invest Dermatol. 2010. № 130. P. 425–429.
№ 130. P. 425–429.
19. Pigors M., Kiritsi D., Cobzaru C., Schwieger-Briel A., Suarez J., et al. TGM5mutations Impact Epidermal Differentiation in Acral Peeling Skin Syndrome // J Invest Dermatol. 2012. № 132. P. 2422–2429.
20. Pigors M., Schwieger-Briel A., Leppert J., Kiritsi D., Kohlhase J., Bruckner-Tuderman L. et al. Molecular Heterogeneity of
21. Epidermolysis Bullosa Simplex: Contribution of EXPH5 Mutations // J Invest Dermatol. 2014. №134. P. 842–845.
22. Yuen W. Y., Pas H. H., Sinke R. J., Jonkman M. F. Junctional
23. Epidermolysis Bullosa of Late Onset Explained by Mutations in
24. COL17A1 // Br J Dermatol. 2011. No. 164. P. 1280–1284.
COL17A1 // Br J Dermatol. 2011. No. 164. P. 1280–1284.
25. Natsuga K., Nishie W., Nishimura M. et al. Loss of Interaction
26. between Plectin and Type XVII Collagen Results in Epidermolysis Bullosa Simplex // Human Mutation. 2017. No. 9. P. 1666–1670. URL: https://onlinelibrary.wiley.com/doi/abs/10.1002/humu.23344 (дата обращения: 13.06.2020).
27. Kamaguchi M., Iwata H., Ujiie H., Natsuga K., Nishie W., Kitagawa Y., Shimizu H. High Expression of Collagen XVII Compensates for its Depletion Induced by Pemphigoid IgG in the Oral Mucosa // Journal of Investigative Dermatology. 2018. № 3. Р. 1707–1715. DOI 10.1016/j.jid.2018.03.002.
28. Альбанова В. И., Гольченко В. А. Лечение буллезного эпидермолиза // Рос. журн. кожных и венерич. болезней. 2013. № 4. С. 21–24.
журн. кожных и венерич. болезней. 2013. № 4. С. 21–24.
29. Сердюкова Е. А., Попов В. В. Лечение врожденного буллезного эпидермолиза у детей // Лекарств. вестн. 2016. Т. 10, № 4 (64). С. 43–47.
30. Goldschneider K. R., Good J., Harrop E. et al. Pain Care for Patients with Epidermolysis Bullosa: Best Care Practice Guidelines // BMC Medicine. 2014. № 12. P. 178.
31. Fujita Y., Abe R., Inokuma D., Sasaki M., Hoshina D., Natsuga K. et al. Bone Marrow Transplantation Restores Epidermal Basement Membrane Protein Expression and Rescues Epidermolysis Bullosa Model Mic // Proc Natl Acad Sci USA. 2010. № 107 (32). P. 14345–14350. DOI 10.1073/pnas.1000044107.
32. Tukaj S., Bieber K., Witte M. et al. Calcitriol Treatment Ameliorates Inflammation and Blistering in Mouse Models of Epidermolysis Bullosa Acquisita // Journal of Investigative Dermatology. 2018. Vol. 138, P. 301–309. DOI 10.1016/j.jid.2017.09.009.
et al. Calcitriol Treatment Ameliorates Inflammation and Blistering in Mouse Models of Epidermolysis Bullosa Acquisita // Journal of Investigative Dermatology. 2018. Vol. 138, P. 301–309. DOI 10.1016/j.jid.2017.09.009.
Генетическая связь между буллезным эпидермолизом и заболеваниями сердца
Актуальность
Международная группа исследователей установила, что генная мутация, ответственная за развитие редкого заболевания кожи, также приводит к серьезному заболеванию сердца.
Речь идет о редком кожном заболевании буллезном эпидермолизе. Буллезный эпидермолиз делает кожу невероятно хрупкой, в связи с чем у пациентов развиваются блистеры и плохо заживающие раны даже при малейшем прикосновении.
Методы и результаты
- Исследователи проанализировали генетические данные более 360 пациентов с буллезным эпидермолизом из разных стран мира.
 Авторы исследования сфокусировали свое внимание на вариантах 21 гена, мутации в которых ответственны за развитие буллезного эпидермолиза.
Авторы исследования сфокусировали свое внимание на вариантах 21 гена, мутации в которых ответственны за развитие буллезного эпидермолиза. - Оказалось, что у 2 пациентов присутствует одинаковая мутация в гене JUP. Любопытно, что у пациентов имели место аналогичные симптомы с раннего детства, включая очень хрупкую кожу, утолщение кожи в области ладоней и подошв и потеря волос, включая область бровей и ресниц. Вы удивитесь, но речь шла о пациентах совершенно разного возраста: 2,5 летний мальчик и 22-летняя женщина. И если у мальчика имели место только симптомы поражения кожи, у женщины также было выявлено поражение сердца – аритмогенная правожелудочковая кардиомиопатия. А это значит, что за мальчиком 2,5 лет необходимо пристально наблюдать, чтобы вовремя выявить кардиальные симптомы и при необходимости имплантировать кардиовертер-дефибриллятор.
- Для аритмогенной правожелудочковой кардиомиопатии характерно замещение здоровой мышечной ткани сердца на фиброзную ткань. Как результат, у пациентов развивается нарушение ритма, сердечная недостаточность и значительно повышается риск внезапной сердечной смерти. Данное заболевание ответственно за 20% случаев внезапной сердечной смерти у пациентов младше 30 лет. Мутация в гене JUP, которая приводит к развитию буллезного эпидермолиза, также ответственна за отложение фиброзной ткани в сердце и аритмогенной правожелудочковой кардиомиопатии.
- Исследователи отмечают, что у пациентов с буллезным эпидермолизом следует мониторировать симптомы, указывающие на патологию сердца.
 Авторы исследования сфокусировали свое внимание на вариантах 21 гена, мутации в которых ответственны за развитие буллезного эпидермолиза.
Авторы исследования сфокусировали свое внимание на вариантах 21 гена, мутации в которых ответственны за развитие буллезного эпидермолиза.  Как результат, у пациентов развивается нарушение ритма, сердечная недостаточность и значительно повышается риск внезапной сердечной смерти. Данное заболевание ответственно за 20% случаев внезапной сердечной смерти у пациентов младше 30 лет. Мутация в гене JUP, которая приводит к развитию буллезного эпидермолиза, также ответственна за отложение фиброзной ткани в сердце и аритмогенной правожелудочковой кардиомиопатии.
Как результат, у пациентов развивается нарушение ритма, сердечная недостаточность и значительно повышается риск внезапной сердечной смерти. Данное заболевание ответственно за 20% случаев внезапной сердечной смерти у пациентов младше 30 лет. Мутация в гене JUP, которая приводит к развитию буллезного эпидермолиза, также ответственна за отложение фиброзной ткани в сердце и аритмогенной правожелудочковой кардиомиопатии.
Источник: Hassan Vahidnezhad, Leila Youssefian, Masoomeh Faghankhani, et al. Arrhythmogenic right ventricular cardiomyopathy in patients with biallelic JUP-associated skin fragility. Scientific Reports, 2020; 10 (1).
Современные особенности клиники, диагностики и терапии больных буллезным эпидермолизом | #01/18
Буллезный эпидермолиз (БЭ) — это группа редких наследственных генетических заболеваний кожи, обусловленных мутациями ряда генов, ответственных за синтез структурных белков кожи. Для заболевания характерна склонность кожи и слизистых оболочек к образованию пузырей, преимущественно на местах незначительного механического воздействия, вследствие нарушения межклеточных связей в эпидермисе или дермоэпидермальном соединении [1, 2]. Наиболее часто пациенты с буллезным эпидермолизом регистрируются в возрасте от 1 до 5 лет. По данным Ю. Ю. Коталевской с соавт., на долю дистрофического варианта приходится больше половины пациентов от общего количества всех форм буллезного эпидермолиза [3]. На сегодняшний день своевременная диагностика и терапия буллезного эпидермолиза остаются одной из главных проблем в медицине. При любой форме буллезный эпидермолиз протекает тяжело, инвалидизация наступает в первые годы течения болезни.
Для заболевания характерна склонность кожи и слизистых оболочек к образованию пузырей, преимущественно на местах незначительного механического воздействия, вследствие нарушения межклеточных связей в эпидермисе или дермоэпидермальном соединении [1, 2]. Наиболее часто пациенты с буллезным эпидермолизом регистрируются в возрасте от 1 до 5 лет. По данным Ю. Ю. Коталевской с соавт., на долю дистрофического варианта приходится больше половины пациентов от общего количества всех форм буллезного эпидермолиза [3]. На сегодняшний день своевременная диагностика и терапия буллезного эпидермолиза остаются одной из главных проблем в медицине. При любой форме буллезный эпидермолиз протекает тяжело, инвалидизация наступает в первые годы течения болезни.
Развитие буллезного эпидермолиза обусловлено мутациями генов, кодирующих структурные белки кожи, которые обеспечивают связь между эпидермисом и дермой. К настоящему времени в 15 генах структурных белков кожи выявлено более 1000 мутаций, способных приводить к развитию различных клинических типов врожденного буллезного эпидермолиза [4–6]. С мутациями связаны нарушения синтеза белков: отсутствие белка, синтез функционально неполноценного белка, синтез белка с нарушениями структуры, облегчающими доступ к белку протеаз, что приводит к его быстрому разрушению. Белками, с которыми связано развитие заболевания, являются кератины 5 и 14, десмоплакин, плакофилин-1, плектин, интегрин α6β4, ламинин 332, коллагены VII и XVII типов, киндлин. Эти белки имеют различную локализацию в коже: в кератиноцитах локализуются кератины 5 и 14, внутри светлой пластинки (lamina lucida) базальной мембраны — интегрин α6β4, ламинин 332, коллаген XVII типа, под темной пластинкой (lamina densa) базальной мембраны — коллаген VII типа, на разных уровнях эпидермиса — киндлин [4, 5].
С мутациями связаны нарушения синтеза белков: отсутствие белка, синтез функционально неполноценного белка, синтез белка с нарушениями структуры, облегчающими доступ к белку протеаз, что приводит к его быстрому разрушению. Белками, с которыми связано развитие заболевания, являются кератины 5 и 14, десмоплакин, плакофилин-1, плектин, интегрин α6β4, ламинин 332, коллагены VII и XVII типов, киндлин. Эти белки имеют различную локализацию в коже: в кератиноцитах локализуются кератины 5 и 14, внутри светлой пластинки (lamina lucida) базальной мембраны — интегрин α6β4, ламинин 332, коллаген XVII типа, под темной пластинкой (lamina densa) базальной мембраны — коллаген VII типа, на разных уровнях эпидермиса — киндлин [4, 5].
Выделяют несколько основных типов буллезного эпидермолиза на основе особенностей механизма образования пузыря и клинической картины: простой буллезный эпидермолиз, пограничный буллезный эпидермолиз, дистрофический буллезный эпидермолиз, синдром Киндлера (разный уровень образования пузырей) [7].
Эпидермолиз буллезный простой
Эпидермолиз буллезный простой характеризуется образованием внутриэпидермальных пузырей в результате дезинтеграции и цитолиза кератиноцитов без признаков рубцевания, атрофии и образования милиумов. Тип наследования аутосомно-доминантный. Первые признаки заболевания обычно проявляются на первом году жизни, иногда могут быть уже при рождении ребенка. На месте легкой травматизации, чаще в области кистей, стоп, спины, локтевых и коленных суставов, затылочной области, на неизмененной коже появляются пузыри различных размеров (от 0,5 до 7 см и более) с плотной покрышкой и прозрачным содержимым. Симптом Никольского отрицательный, акантолитические клетки в содержимом пузыря отсутствуют. Через несколько днем пузыри вскрываются, образуя эрозии, покрывающиеся корками и быстро эпителизирующиеся, не оставляя рубцовых изменении кожи или атрофии. Пузырей обычно больше в теплый период года при выраженном гипергидрозе. С возрастом поражения локализуются в основном на конечностях, особенно на стопах и кистях, чему способствует большая травматизация этих участков кожи, тесная, плохо подобранная обувь, а также на участках тесного прилегания одежды. Пузыри появляются на протяжении всей жизни, но в постпубертатный период их количество уменьшается. Слизистые оболочки, ногти не поражаются или их изменения минимальны. Общее состояние больного не изменяется. Возможна пренатальная диагностика этой формы заболевания по высокому содержанию в сыворотке крови беременной α-фетопротеина во II триместре. Эпидермолиз буллезный простой летний Вебера–Коккейна — абортивная локализованная форма эпидермолиза буллезного простого. Характеризуется образованием пузырей на коже кистей и стоп лишь в летнее время года при выраженном ладонно-подошвенном гипергидрозе [8, 12].
Пузыри появляются на протяжении всей жизни, но в постпубертатный период их количество уменьшается. Слизистые оболочки, ногти не поражаются или их изменения минимальны. Общее состояние больного не изменяется. Возможна пренатальная диагностика этой формы заболевания по высокому содержанию в сыворотке крови беременной α-фетопротеина во II триместре. Эпидермолиз буллезный простой летний Вебера–Коккейна — абортивная локализованная форма эпидермолиза буллезного простого. Характеризуется образованием пузырей на коже кистей и стоп лишь в летнее время года при выраженном ладонно-подошвенном гипергидрозе [8, 12].
Эпидермолиз буллезный соединительный
Эпидермолиз буллезный соединительный характеризуется образованием подэпидермальных пузырей за счет поражения lamina lucida эпидермодермального соединения, расположенной между плазматической мембраной базальных кератиноцитов и базальной мембраной кожи, и развитием атрофических изменений кожи в очагах поражения. Возможна пренатальная диагностика с помощью биопсии кожи 18-недельного плода на основании выявления указанных изменений. Тип наследования аутосомно-рецессивный.
Тип наследования аутосомно-рецессивный.
Процесс характеризуется появлением пузырей и эрозий уже при рождении ребенка или вскоре после него. В течение нескольких дней процесс генерализуется. Основная локализация высыпаний — кожа груди, головы, слизистые оболочки рта, гортани, трахеи. Хотя кожа кистей и стоп не изменена, ногтевые пластинки дистрофичны, развиваются анонихия, акроостеолиз. Образующиеся на месте пузырей эрозивные поверхности заживают медленно, оставляя участки атрофии кожи. Рубцов и милиумов нет. Многие дети умирают в первые месяцы жизни от сепсиса, анемии [8].
Эпидермолиз буллезный дистрофический
Эпидермолиз буллезный дистрофический характеризуется образованием пузырей вследствие дерматолиза — гибели коллагеновых фибрилл в сосочковом слое дермы ниже lamina densa. Формируются эрозивно-язвенные поверхности, заживающие рубцами, характерны также образование милиумов, изменение ногтей, волос, зубов и другие аномалии.
Эпидермолиз буллезный дистрофический рецессивный генерализованный
Эпидермолиз буллезный дистрофический рецессивный генерализованный (эпидермолиз буллезный дистрофический полидиспластический) отличается образованием пузырей в сосочковом слое дермы и в результате дерматолиза — лизиса коллагеновых фибрилл с фагоцитозом их макрофагами и разрушениями ниже lamina densa. Патологический процесс связывают с увеличением уровня и активности фермента коллагеназы, разрушающей основной компонент опорных коллагеновых фибрилл — коллаген VII (коллагенолиз). Возможна пренатальная диагностика болезни по результатам биопсии кожи плода на 21-й неделе развития и выявления описанных ранее изменений. Первые признаки заболевания появляются уже при рождении (60% больных) или в первые недели жизни. Крупные пузыри, нередко с гемморрагическим содержимым, возникают спонтанно на любом участке кожного покрова и слизистых оболочек. Обширные длительно не заживающие эрозивно-язвенные поверхности, образующиеся при их вскрытии, затрудняют уход и вскармливание новорожденных. Симптом эпидермальной отслойки положительный. На эрозивно-язвенных, нередко кровоточащих, болезненных участках развиваются вегетации. Заживление их происходит медленно, с формированием уродствующих атрофических рубцов. Рубцовые изменения пищевода, глотки, слизистой оболочки рта могут затруднять прием пищи, облитерировать выводные протоки слюнных желез, ограничивать подвижность языка и привести к развитию лейкоплакии. Поражения глаз в виде эрозивно-язвенного кератита с последующим рубцеванием приводят к потере зрения, рубцовому эктропиону, облитерации протоков слезных желез. Наблюдаются также акроцианоз, склеродермоподобные изменения кожи кистей, стоп с формированием сгибательных контрактур суставов, акроостеолизом и характерной деформацией кистей по типу «варежки» в результате срастания и деформации пальцев. Характерна также дистрофия ногтей, волос, зубов. Возможны нарушения эндокринной (гипофункция щитовидной железы, гипофиза), нервной (эпилепсия, отставание умственного развития) систем. Отмечается высокая летальность в раннем детском возрасте от сепсиса, анемии, нарушения питания, в более старшем возрасте — от злокачественных новообразований кожи, пищевода, органов полости рта [8].
Поражения глаз в виде эрозивно-язвенного кератита с последующим рубцеванием приводят к потере зрения, рубцовому эктропиону, облитерации протоков слезных желез. Наблюдаются также акроцианоз, склеродермоподобные изменения кожи кистей, стоп с формированием сгибательных контрактур суставов, акроостеолизом и характерной деформацией кистей по типу «варежки» в результате срастания и деформации пальцев. Характерна также дистрофия ногтей, волос, зубов. Возможны нарушения эндокринной (гипофункция щитовидной железы, гипофиза), нервной (эпилепсия, отставание умственного развития) систем. Отмечается высокая летальность в раннем детском возрасте от сепсиса, анемии, нарушения питания, в более старшем возрасте — от злокачественных новообразований кожи, пищевода, органов полости рта [8].
Эпидермолиз буллезный дистрофический доминантный
Эпидермолиз буллезный дистрофический доминантный (эпидермолиз буллезный дистрофический гиперпластический) характеризуется образованием пузырей в дерме (дерматолиз) ниже lamina densa за счет гибели опорных коллагеновых фибрилл; возможна пренатальная диагностика (по аналогии с дистрофическим полидиспластическим буллезным эпидермолизом). Тип наследования аутосомно-доминантный. Первые проявления болезни появляются в раннем детском возрасте или несколько позднее (4–10 лет). Пузыри возникают после незначительной травмы, чаще в области конечностей. Они напряженные, плотные, с серозным или геморрагическим содержимым: вскрываясь, образуют эрозивно-язвенные поверхности, заживающие медленно с образованием мягкой или келоидоподобной рубцовой атрофии вначале розового, затем белого цвета. В области суставов на месте пузырей формируются обширные поля поражения в виде рубцовой ткани с множеством эпидермальных кист (милиумы). Симптом эпидермальной отслойки положительный. Ногти, вовлеченные в процесс, утолщены, дистрофичны. Слизистые оболочки поражаются редко. Волосы, зубы и общее развитие обычно не изменяются, однако часто отмечается ассоциация с ихтиозом, фолликулярным кератозом, гипертрихозом. Диагноз буллезного эпидермолиза основывается на клинических и гистологических данных. Возможна пренатальная диагностика заболевания. Дифференциальный диагноз в раннем детском возрасте проводят с эпидермолитическим ихтиозом, при котором доминирует кератоз; эпидемической пузырчаткой новорожденных, для которой характерно острое начало с лихорадкой, интоксикацией и воспалительными пузырями в результате некротических процессов в эпидермисе, вызванных стафилококком.
Тип наследования аутосомно-доминантный. Первые проявления болезни появляются в раннем детском возрасте или несколько позднее (4–10 лет). Пузыри возникают после незначительной травмы, чаще в области конечностей. Они напряженные, плотные, с серозным или геморрагическим содержимым: вскрываясь, образуют эрозивно-язвенные поверхности, заживающие медленно с образованием мягкой или келоидоподобной рубцовой атрофии вначале розового, затем белого цвета. В области суставов на месте пузырей формируются обширные поля поражения в виде рубцовой ткани с множеством эпидермальных кист (милиумы). Симптом эпидермальной отслойки положительный. Ногти, вовлеченные в процесс, утолщены, дистрофичны. Слизистые оболочки поражаются редко. Волосы, зубы и общее развитие обычно не изменяются, однако часто отмечается ассоциация с ихтиозом, фолликулярным кератозом, гипертрихозом. Диагноз буллезного эпидермолиза основывается на клинических и гистологических данных. Возможна пренатальная диагностика заболевания. Дифференциальный диагноз в раннем детском возрасте проводят с эпидермолитическим ихтиозом, при котором доминирует кератоз; эпидемической пузырчаткой новорожденных, для которой характерно острое начало с лихорадкой, интоксикацией и воспалительными пузырями в результате некротических процессов в эпидермисе, вызванных стафилококком.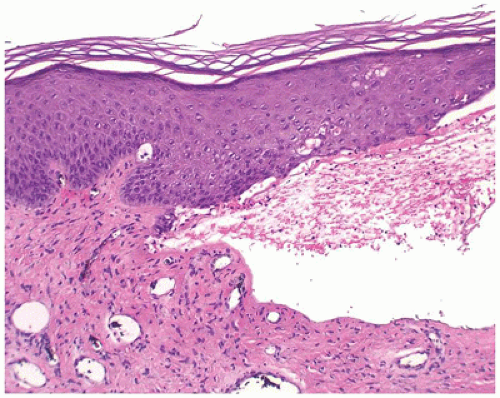 У детей более старшего возраста некоторые формы БЭ дифференцируют от доброкачественного буллезного пемфигоида, который отличается линеарным отложением IgА вдоль назальной мембраны. Антитела к lamina densa, VII типу коллагена, пемфигоидные и др. помогают установить характер дефекта и уточнить диагностику [8, 9].
У детей более старшего возраста некоторые формы БЭ дифференцируют от доброкачественного буллезного пемфигоида, который отличается линеарным отложением IgА вдоль назальной мембраны. Антитела к lamina densa, VII типу коллагена, пемфигоидные и др. помогают установить характер дефекта и уточнить диагностику [8, 9].
На основании только анамнестических и клинических данных не всегда представляется возможным установить диагноз БЭ. У детей старшего возраста в диагностике помогает сбор анамнеза (начало заболевания, значение механического фактора в развитии пузырей и эрозий, постоянно прогрессирующее течение, последовательность развития симптомов, наследственная отягощенность). Обнаружение определенных специфических клинических признаков некоторых форм БЭ может указать на данную патологию, но в большинстве случаев для уточнения диагноза требуется проведение лабораторной диагностики. Традиционное патоморфологическое исследование биоптата кожи не позволяет различить клинические формы БЭ между собой, но при определенном опыте можно отличить по уровню образования пузыря от истинной акантолитической пузырчатки. После исключения всех других возможных заболеваний для подтверждения диагноза заподозренного БЭ и установления его группы могут быть использованы дополнительные методы диагностики: иммунофлюоресцентное антигенное картирование (ИАК), трансмиссионная электронная микроскопия (ТЭМ) и генетический анализ (молекулярная или ДНК-диагностика) [9].
После исключения всех других возможных заболеваний для подтверждения диагноза заподозренного БЭ и установления его группы могут быть использованы дополнительные методы диагностики: иммунофлюоресцентное антигенное картирование (ИАК), трансмиссионная электронная микроскопия (ТЭМ) и генетический анализ (молекулярная или ДНК-диагностика) [9].
Трансмиссионная электронная микроскопия
До начала использования в практике иммунофлюоресцентного анализа ТЭМ являлась «золотым» стандартом в диагностике БЭ, так как позволяла определить уровень образования пузыря и обнаружить ультраструктурные изменения в коже и непосредственно в поврежденных структурах, таких как кератиновые филаменты, десмосомы, полудесмосомы, крепящие (якорные) филаменты, крепящие (якорные) фибриллы, которые при определенных формах БЭ могут отсутствовать, либо содержаться в коже в недостаточном количестве, либо иметь дефектную структуру [9].
Иммунофлюоресцентное антигенное картирование
Иммунофлюоресцентное антигенное картирование было впервые описано в 1981 г. В основе метода лежит последовательная обработка препарата кожи пациента первичными антителами к структурным белкам кожи и вторичными антителами, мечеными флюоресцирующей меткой, которые связываются с первичными антителами. В связи с тем, что известно большое количество белков, дефекты которых приводят к развитию БЭ (кератины 5 и 14, плектин, плакофилин-1, десмоплакин, ламинин 332, интегрин α6β4, коллагены VII и XVII типов, киндлин-1), необходимо определять экспрессию каждого из них. Поэтому для проведения исследования методом ИАК из полученного биоптата кожи изготавливают несколько гистологических препаратов [9–11].
В основе метода лежит последовательная обработка препарата кожи пациента первичными антителами к структурным белкам кожи и вторичными антителами, мечеными флюоресцирующей меткой, которые связываются с первичными антителами. В связи с тем, что известно большое количество белков, дефекты которых приводят к развитию БЭ (кератины 5 и 14, плектин, плакофилин-1, десмоплакин, ламинин 332, интегрин α6β4, коллагены VII и XVII типов, киндлин-1), необходимо определять экспрессию каждого из них. Поэтому для проведения исследования методом ИАК из полученного биоптата кожи изготавливают несколько гистологических препаратов [9–11].
Метод используется для определения уровня образования пузыря (внутриэпидермально, внутри светлой пластинки базальной мембраны, под плотной пластинкой базальной мембраны). ИАК позволяет также определить, дефицит какого из связывающих структуры дермы и эпидермиса белков наблюдается. ИАК определяет экспрессию структурных белков в зоне дермоэпидермального соединения, то есть их присутствие, снижение или отсутствие их экспрессии. Для исследований методом ИАК используют первичные и вторичные моноклональные и поликлональные антитела. Этот метод диагностики более распространен, легче выполним и дешевле по сравнению с ТЭМ. Для правильной постановки диагноза методом ИАК нужно иметь достаточный опыт получения биообразцов кожи, причем одним из требований к получению биообразцов является наличие свежего (не более 24 часов) пузыря на коже. Пузырь может быть как появившимся спонтанно, так и индуцированным намеренно, например, с помощью трения кожи. Биопсию проводят на границе видимо здоровой кожи и свежего пузыря или в зоне трения (через 30 мин после его окончания). Полученный биоптат сразу же подвергается заморозке жидким азотом или помещается в физиологический раствор (0,9% NaCl) на срок не более 24 часов. Для более продолжительного хранения используют специальную транспортную среду Michel. В таком состоянии биообразцы кожи могут сохраняться в течение нескольких недель [12].
Для исследований методом ИАК используют первичные и вторичные моноклональные и поликлональные антитела. Этот метод диагностики более распространен, легче выполним и дешевле по сравнению с ТЭМ. Для правильной постановки диагноза методом ИАК нужно иметь достаточный опыт получения биообразцов кожи, причем одним из требований к получению биообразцов является наличие свежего (не более 24 часов) пузыря на коже. Пузырь может быть как появившимся спонтанно, так и индуцированным намеренно, например, с помощью трения кожи. Биопсию проводят на границе видимо здоровой кожи и свежего пузыря или в зоне трения (через 30 мин после его окончания). Полученный биоптат сразу же подвергается заморозке жидким азотом или помещается в физиологический раствор (0,9% NaCl) на срок не более 24 часов. Для более продолжительного хранения используют специальную транспортную среду Michel. В таком состоянии биообразцы кожи могут сохраняться в течение нескольких недель [12].
Генетический анализ (молекулярная или ДНК-диагностика)
Генетический анализ (молекулярная или ДНК-диагностика) является оптимальным методом для определения типа наследования и специфических мутаций, имеющихся у больных БЭ, а также наиболее точным методом для верификации различных клинических форм простого, пограничного и дистрофического БЭ. Поиск известных мутаций является первичным диагностическим подходом, особенно в семьях больных БЭ с аутосомно-доминантным типом наследования, в силу того, что данные мутации, как правило, идентичны в семье, страдающей данным заболеванием. Также имеются данные, что поиск неизвестных мутаций может быть осуществлен с использованием ДНК-амплификационного метода и секвенированием РНК дефектных генов [9].
Поиск известных мутаций является первичным диагностическим подходом, особенно в семьях больных БЭ с аутосомно-доминантным типом наследования, в силу того, что данные мутации, как правило, идентичны в семье, страдающей данным заболеванием. Также имеются данные, что поиск неизвестных мутаций может быть осуществлен с использованием ДНК-амплификационного метода и секвенированием РНК дефектных генов [9].
Пренатальная диагностика
Как известно, до настоящего времени не разработано эффективных методов терапии БЭ, поэтому особенно важное значение на стадии планирования семьи, находящейся в группе риска наследования данной патологии, приобретает пренатальная диагностика и генетическое консультирование. Первое сообщение о пренатальной диагностике одной из наиболее тяжелых форм БЭ (летального генерализованного БЭ Герлитца) было опубликовано еще в 1980 г. [13].
Усовершенствование молекулярных методов диагностики позволило проводить пренатальную диагностику на основании анализа фетальной ДНК, которую можно получить из амниотической жидкости и/или ворсинок хориона. Биопсия ворсин хориона и амниоцентез позволяют существенно снизить риск преждевременного прерывания беременности по сравнению с фетоскопией, а также провести данный вид диагностики в I триместре (до 11 недель беременности). Дородовая диагностика возможна только в том случае, когда ее планируют заранее. До наступления беременности нужно установить точно генетический дефект у болеющего члена семьи. Если дефектный ген уже известен, то поиск такой же поломки у плода занимает всего несколько дней, что создает возможность проведения аборта в безопасные для беременной женщины сроки. Трудностью в диагностике может являться мозаицизм, при котором у ребенка могут присутствовать две и более генетически различные популяции клеток [14, 15].
Биопсия ворсин хориона и амниоцентез позволяют существенно снизить риск преждевременного прерывания беременности по сравнению с фетоскопией, а также провести данный вид диагностики в I триместре (до 11 недель беременности). Дородовая диагностика возможна только в том случае, когда ее планируют заранее. До наступления беременности нужно установить точно генетический дефект у болеющего члена семьи. Если дефектный ген уже известен, то поиск такой же поломки у плода занимает всего несколько дней, что создает возможность проведения аборта в безопасные для беременной женщины сроки. Трудностью в диагностике может являться мозаицизм, при котором у ребенка могут присутствовать две и более генетически различные популяции клеток [14, 15].
Преимплантационная генетическая диагностика
Преимплантационная генетическая диагностика является перспективным альтернативным методом наряду с вышеперечисленными, кроме того, метод может быть частью экстракорпорального оплодотворения, выявляя тем самым различные отклонения развития эмбриона еще на стадии бластоцисты. Суть данного метода заключается в оплодотворении овоцита (незрелой яйцеклетки) in vitro, что ведет к образованию эмбриона. Когда эмбрион достигает стадии 8- или 12-клеточного субъекта, производится отщепление одной клетки для дальнейшего генетического анализа. Если генетический анализ указывает на нормальный генотип — эмбрион подсаживают в матку, иначе его утилизируют [16].
Суть данного метода заключается в оплодотворении овоцита (незрелой яйцеклетки) in vitro, что ведет к образованию эмбриона. Когда эмбрион достигает стадии 8- или 12-клеточного субъекта, производится отщепление одной клетки для дальнейшего генетического анализа. Если генетический анализ указывает на нормальный генотип — эмбрион подсаживают в матку, иначе его утилизируют [16].
Неинвазивные методы пренатальной диагностики
Все вышеперечисленные методы пренатальной диагностики являются инвазивными и в той или иной степени могут повлиять на развитие плода и на течение беременности. В связи с этим идет постоянный поиск новых методов, снижающих риск инвазивного воздействия [9].
Среди таких методов можно выделить метод ультразвуковой диагностики, который позволяет визуализировать некоторые особенности развития плода при ограниченном количестве тяжелых патологий, а также другие структуры содержимого матки, которые могут быть идентификаторами некоторых врожденных заболеваний. К таким показателям можно отнести особенности фенотипа плодного яйца: размеры носовой кости, воротниковой области, желточного мешка [9].
К таким показателям можно отнести особенности фенотипа плодного яйца: размеры носовой кости, воротниковой области, желточного мешка [9].
Генетическое консультирование лучше проводить дерматологом или генетиком, специализирующимся на БЭ. В конечном итоге диагноз ставится на основании клинического фенотипа, способа наследования, а также, если возможно, провести мутационный анализ пробанда.
В периоде новорожденности наблюдение и симптоматическая терапия за больными врожденным буллезным эпидермолизом (ВБЭ) проводится в условиях отделения интенсивной терапии педиатрического стационара. Основные принципы терапии не отличаются от таковых у взрослых лиц. Лекарственная терапия проводится с учетом возрастных ограничений к назначению лекарственных препаратов. Профилактические прививки противопоказаны только в период нарушения общего состояния ребенка. Беременность у больных ВБЭ обычно протекает без осложнений. При планировании беременности необходимо провести лечение сопутствующих заболеваний, в том числе очагов хронической инфекции, хирургическое устранение контрактур и псевдосиндактилии. Во избежание травмирования принимают меры предосторожности при взятии крови, вагинальном исследовании, пальпации, УЗИ. Основным в период беременности является наружное лечение [7].
Во избежание травмирования принимают меры предосторожности при взятии крови, вагинальном исследовании, пальпации, УЗИ. Основным в период беременности является наружное лечение [7].
Больным буллезным эпидермолизом необходимо избегать физических нагрузок, связанных с повышением потоотделения, травмоопасных ситуаций, резких движений. Перевязочные материалы, одежда, закрытая обувь позволяют свести к минимуму травмирование кожи. При хорошем самочувствии и отсутствии высыпаний на коже допустимо плавание.
Тяжелые подтипы БЭ, а также нетяжелые, но протекающие с поражением полости рта, требуют особого внимания к питанию. Диета должна быть механически, термически и химически щадящей (протертой и полужидкой, не горячей). Питание особенно важно для больных с большой площадью поражения кожи, так как они теряют питательные вещества и влагу с тканевой жидкостью, необходимой для заживления ран и борьбы с инфицированием. Питание больных должно быть богато белками, углеводами, жирами, а также содержать витамины, минералы, пищевые волокна и большое количество жидкости. При однообразной диете и недостаточном питании потребление витаминов и минералов с пищей ограничено, поэтому рекомендуется дополнительно принимать поливитаминно-минеральные комплексы. При наличии эрозий во рту, дисфагии и сужении пищевода назначают жидкие комплексы. В их составе особенно важны витамины А, группа В, С, D и Е, из минералов — железо, цинк, селен и кальций: витамин А (ретинола пальмитат), масляный раствор 100 000 МЕ/мл перорально на ночь по 10–30 капель (в зависимости от возраста и веса пациента) в течение 2 месяцев. Курсы терапии можно повторять с интервалом в 3 месяца; витамин С (аскорбиновая кислота) 100 мг перорально после еды 3–4 раза в сутки в течение 2 недель. При большом количестве пузырей и эрозий пациент нуждается в восполнении теряемой жидкости [7, 17].
При однообразной диете и недостаточном питании потребление витаминов и минералов с пищей ограничено, поэтому рекомендуется дополнительно принимать поливитаминно-минеральные комплексы. При наличии эрозий во рту, дисфагии и сужении пищевода назначают жидкие комплексы. В их составе особенно важны витамины А, группа В, С, D и Е, из минералов — железо, цинк, селен и кальций: витамин А (ретинола пальмитат), масляный раствор 100 000 МЕ/мл перорально на ночь по 10–30 капель (в зависимости от возраста и веса пациента) в течение 2 месяцев. Курсы терапии можно повторять с интервалом в 3 месяца; витамин С (аскорбиновая кислота) 100 мг перорально после еды 3–4 раза в сутки в течение 2 недель. При большом количестве пузырей и эрозий пациент нуждается в восполнении теряемой жидкости [7, 17].
В настоящее время пренатальная диагностика является лучшим способ профилактики тяжелых наследственных заболеваний. Она возможна только при планировании беременности задолго до ее наступления. Медико-генетическое консультирование семьи проводится с целью оценки риска появления больного ребенка в семье, информирования семьи о риске развития наследственного заболевания, о возможных диагностических и терапевтических методах. Показания для консультирования — предыдущее рождение ребенка с ВБЭ, наличие заболевания у одного из родителей, установленное или подозреваемое заболевание в семье. Генетический анализ крови и кожи больного позволяет уточнить тип и подтип ВБЭ, а также обнаружить мутацию соответствующего гена. При наступлении беременности в сроки 10–12 недель проводят биопсию ворсин хориона, в которых ведется поиск уже известной мутации. Быстрое получение результатов (в течение 3–4 дней после взятия материала) позволяет принять решение о прерывании беременности своевременно.
Показания для консультирования — предыдущее рождение ребенка с ВБЭ, наличие заболевания у одного из родителей, установленное или подозреваемое заболевание в семье. Генетический анализ крови и кожи больного позволяет уточнить тип и подтип ВБЭ, а также обнаружить мутацию соответствующего гена. При наступлении беременности в сроки 10–12 недель проводят биопсию ворсин хориона, в которых ведется поиск уже известной мутации. Быстрое получение результатов (в течение 3–4 дней после взятия материала) позволяет принять решение о прерывании беременности своевременно.
У пациента с БЭ профилактические меры в отношении появления пузырей на коже и слизистых оболочках включают ограничение возможности травмирования кожи (одежда, диета, особенности ухода, неадгезивные повязки, наружные средства, уход за полостью рта). Таким образом, профилактика развития осложнений, диспансеризация, периодический контроль лабораторных показателей для выявления и контроля анемии, полный осмотр пациентов с целью раннего выявления злокачественных опухолей кожи, своевременное лечение зубов будут способствовать существенному облегчению и улучшению качаства жизни пациентов.
Литература
- Lara-Corrales I., Mellerio J. E., Martinez A. E. et al. Dilated cardiomyopathy in epidermolysis bullosa: a retrospective, multicenter study // Pediatr Dermatol. 2010; 27 (3): p. 238–243.
- Гараева З. Ш., Юсупова Л. А., Мавлютова Г. И., Юнусова Е. И., Мирзина Д. Р. Буллезный эпидермолиз. Сборник материалов Всероссийской научно-практической конференции с международным участием «Казанские дерматологические чтения: синтез науки и практики». Казань, 2016. С. 8–20.
- Коталевская Ю. Ю., Кропачева В. В., Марычева Н. М. Буллезный эпидермолиз — состояние проблемы в России. Материалы I Евразийской Конференции по редким заболеваниям и редким лекарствам и III Всероссийской Конференции по редким заболеваниям и редко применяемым медицинским технологиям «Дорога жизни». М., 2012. С. 15–19.
- Mitsuhashi Y., Hashimoto I. Genetic abnormalities and clinical classification of epidermolysis bullosa // Arch Dermatol Res. 2003; 295 (Suppl 1.): 29–33.
- Uitto J., Richard G. Progress in epidermolysis bullosa: genetic classification and clinical implications // Am J Med Genet C Semin Med Genet. 2004; 131 C (1): 61–74.
- Юсупoвa Л. A. Иммунопатология хронических дерматозов. Казань: НБ КГМА, 2017. 108 с.
- Федеральные клинические рекомендации. Дерматовенерология 2015: Болезни кожи. Инфекции, передаваемые половым путем. 5-е изд., перераб. и доп. М.: Деловой экспресс. 2016. 768 с.
- Иванов О. И. Кожные и венерические болезни: Шико. М., 2006. 133–137 с.
- Альбанова В. И., Чикин В. В., Епишев Р. В. К вопросу о диагностике врожденного буллезного эпидермолиза // Вестник дерматологии и венерологии. 2014. № 3. С. 53–59.
- Pohla-Gubo G., Cepeda-Valdes R., Hintner H. Immunofluorescence mapping for the diagnosis of epidermolysis bullosa // Dermatol Clin. 2010; 28: p. 201–210.
- Cepeda-Valdés R. , Pohla-Gubo G., Borbolla-Escoboza J. R. et al. Immunofluorescence mapping for diagnosis of congenital epidermolysis bullosa // Actas Dermosifiliogr. 2010; 101 (8): p. 673–682.
- Intong L. R., Murrell D. F. How to take skin biopsies for epidermolysis bullosa // Dermatol Clin. 2010; 28: p. 197–200.
- Rodeck C. H., Eady R. A., Gosden C. M. Prenatal diagnosis of epidermolysis bullosa letalis // Lancet. 1980; 1: p. 949–952.
- Pasmooij A. M., Pas H. H., Bolling M. C., Jonkman M. F. Revertant mosaicism in junctional epidermolysis bullosa due to multiple correcting second-site mutations in LAMB3 // J Clin Invest. 2007; 117 (5): p. 1240–1248.
- Pasmooij A. M., Pas H. H., Deviaene F. C. et al. Multiple correcting COL17 A1 mutations in patients with revertant mosaicism of epidermolysis bullosa // Am J Hum Genet. 2005; 77 (5): p. 727–740.
- Renwick P., Ogilvie C. M. Preimplantation genetic diagnosis for monogenic diseases: overview and emerging issues // Expert Rev Mol Diagn. 2007; 7: p. 33–43.
- Boeira V. L., Souza E. S., Rocha Bde O. et al. Inherited epidermolysis bullosa: clinical and therapeutic aspects // An Bras Dermatol 2013; 88 (2): p. 185–198.
 2003; 295 (Suppl 1.): 29–33.
2003; 295 (Suppl 1.): 29–33. 2007; 7: p. 33–43.
2007; 7: p. 33–43. Л. А. Юсупова*, 1, доктор медицинских наук, профессор
Е. И. Юнусова*, кандидат медицинских наук
З. Ш. Гараева*, кандидат медицинских наук
Г. И. Мавлютова*, кандидат медицинских наук
М. А. Морозова**
* ГБОУ ДПО КГМА МЗ РФ, Казань
** ГАУЗ РККВД, Казань
1 Контактная информация: [email protected]
Современные особенности клиники, диагностики и терапии больных буллезным эпидермолизом/ Л. А. Юсупова, Е. И. Юнусова, З. Ш. Гараева, Г. И. Мавлютова, М. А. Морозова
Для цитирования: Лечащий врач № 1/2018; Номера страниц в выпуске: 71-74
Теги: заболевания кожи, генетические мутации, белки
Целесообразность исследования состава тела с целью оценки и мониторинга нутритивного статуса у детей с дистрофической формой врожденного буллезного эпидермолиза | Макарова
1. Fine JD, Bruckner-Tuderman L, Eady RA, et al. Inheritedepidermolysis bullosa: updated recommendations on diagnosisand classification. J Am Acad Dermatol. 2014;70(6):1103–1126.doi: 10.1016/j.jaad.2014.01.903.
Fine JD, Bruckner-Tuderman L, Eady RA, et al. Inheritedepidermolysis bullosa: updated recommendations on diagnosisand classification. J Am Acad Dermatol. 2014;70(6):1103–1126.doi: 10.1016/j.jaad.2014.01.903.
2. Буллезный эпидермолиз / Под ред. Дж.-Д.Файна, Х. Хинтнера. — М.; 2014. — 357 c.
3. Watkins J. Diagnosis, treatment and management of epidermolysis bullosa. Br J Nurs. 2016;25(8):428–431. doi: 10.12968/ bjon.2016.25.8.428.
4. Макарова С.Г., Намазова-Баранова Л.С., Мурашкин Н.Н., и др. Коррекция нутритивного статуса в комплексной терапии детей, страдающих дистрофической формой врожденного буллезного эпидермолиза диагностика в педиатрии // Педиатрическая фармакология. — 2016. — Т.13. — №6 — С. 577–587. doi: 10.15690/pf.v13i6.1672.
5. Fox AT, Alderdice F, Atherton DJ. Are children with recessive dys-trophic epidermolysis bullosa of low birthweight? Pediatr Dermatol. 2003;20(4):303–306. doi: 10.1046/j.1525-1470.2003.20404.x.
Fox AT, Alderdice F, Atherton DJ. Are children with recessive dys-trophic epidermolysis bullosa of low birthweight? Pediatr Dermatol. 2003;20(4):303–306. doi: 10.1046/j.1525-1470.2003.20404.x.
6. Wells JC, Fewtrell MS. Is body composition important for paediatricians? Arch Dis Child. 2008;93(2):168–172. doi: 10.1136/ adc.2007.115741.
7. Wrottesley SV, Pisa PT, Micklesfield LK, et al. A comparison of body composition estimates using dual-energy X-ray absorptiometry and air-displacement plethysmography in South African neonates. Eur J Clin Nutr. 2016;70(11):1254–1258. doi: 10.1038/ejcn.2016.91.
8. Беляева И.А., Намазова-Баранова Л.С., Тарзян Э.О., и др. Особенности физического развития и состава тканей тела недоношенных детей , получавших различные виды вскармливания (при выписке из стационара 2-го этапа выхаживания) // Вестник Российской академии медицинских наук.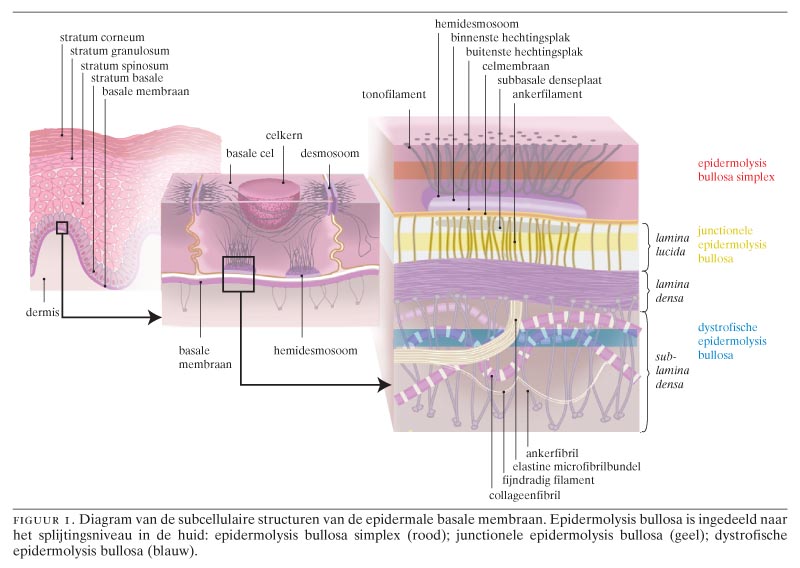 — 2014. — Т.69. — №5–6 — С. 71–80. doi: 10.15690/vramn. v69i5-6.1047.
— 2014. — Т.69. — №5–6 — С. 71–80. doi: 10.15690/vramn. v69i5-6.1047.
9. Barbosa-Silva MC, Barros AJ. Bioelectrical impedance analysis in clinical practice: a new perspective on its use beyond body composition equations. Curr Opin Clin Nutr Metab Care. 2005;8(3):311– 317. doi: 10.1097/01.mco.0000165011.69943.39.
10. Böhm A, Heitmann BL. The use of bioelectrical impedance analysis for body composition in epidemiological studies. Eur J Clin Nutr. 2013;67 Suppl 1:S79–85. doi: 10.1038/ejcn.2012.168.
11. Николаев Д.В., Руднев С.Г. Биоимпедансный анализ: основы метода, протокол обследования и интерпретация результатов // Спортивная медицина: наука и практика. — 2012. — №2 — С. 29–36.
12. Николаев Д.В., Смирнов А.В., Бобринская И.Г., Руднев С.Г. Биоимпедансный анализ состава тела человека. — М.: Наука; 2009. — 392 с.
Николаев Д.В., Смирнов А.В., Бобринская И.Г., Руднев С.Г. Биоимпедансный анализ состава тела человека. — М.: Наука; 2009. — 392 с.
13. Makarova S, Namazova-Baranova L, Murashkin N, et al. Nutritional status in patients with different forms of epidermolysis bullosa. Arch Dis Child. 2017;102(Suppl 2):A49–A50. doi: 10.1136/ archdischild-2017-313273.127.
14. Fields DA, Gunatilake R, Kalaitzoglou E. Air displacement plethysmography: cradle to grave. Nutr Clin Pract. 2015;30(2):219–226. doi: 10.1177/0884533615572443.
15. Kyle UG, Genton L, Pichard C. Low phase angle determined by bioelectrical impedance analysis is associated with malnutrition and nutritional risk at hospital admission. Clin Nutr. 2013;32(2):294– 299. doi: 10.1016/j.clnu.2012.08.001.
16. Kyle UG, Bosaeus I, De Lorenzo AD, et al. Bioelectrical impedance analysis, part II: utilization in clinical practice. Clin Nutr. 2004;23(6):1430–1453. doi: 10.1016/j.clnu.2004.09.012.
Kyle UG, Bosaeus I, De Lorenzo AD, et al. Bioelectrical impedance analysis, part II: utilization in clinical practice. Clin Nutr. 2004;23(6):1430–1453. doi: 10.1016/j.clnu.2004.09.012.
17. Приказ Минздрава России от 15.11.2012 №920н «Об утверждении Порядка оказания медицинской помощи населению по профилю «диетология» (зарегистрировано в Минюсте России 17.04.2013 №28162).
Аутосомно-доминантный буллезный эпидермолиз дистрофического типа у новорожденного | Захарова
1. Швед А.Д., Туровец А.Н. Буллезный эпидермолиз: подходы генной и клеточной терапии. Клеточная трансплантология и тканевая инженерия 2011; VI(4): 21–25. [Shved A.D., Tourovets A.N. Еpidermolysis bullosa: approaches of gene and cell therapy. Kletochnaya transplantologiya i tkanevaya inzheneriya 2011; VI(4): 21–25. (in Russ)]
2. Новиков П.В. Правовые аспекты редких (орфанных) заболеваний в России и в мире. Медицина 2013; 4: 53–73. [Novikov P.V. Legal Issues Relating to Rare (Orphan) Diseases in Russia and in the world. Meditsina 2013; 4: 53–73. (in Russ)]
3. Альбанова В.И., Гольчено В.А. Наследственный буллезный эпидермолиз: современные представления об этиологии и патогенезе. Российский журнал кожных и венерических болезней 2013; 2: 15–20. [Albanova V.I., Golchenko V.A. Hereditary bullous epidermolysis. Modern concepts of the etiology and pathogenesis. Rossiiskii zhurnal kozhnykh i venericheskikh boleznei 2013; 2: 15–20. (in Russ)]
4. Федеральные клинические рекомендации по ведению больных врожденным буллезным эпидермолизом. Российское общество дерматовенерологов и косметологов. Москва, 2015 // http://www.pediatrrussia.ru/sites/default/ files/file/kr_vbe.pdf. [Federal clinical guidelines for the management of patients with congenital epidermolysis bullosa. Rossijskoe obshhestvo dermatovenerologov i kosmetologov. Moscow, 2015 // http://www.pediatrrussia.ru/sites/default/ files/file/kr_vbe.pdf. (in Russ)]
5. Намазова-Баранова Л.С., Торшхоева Р.М., Беляева И.А. Уход за кожей новорожденного ребенка. Методические рекомендации. М., 2016; 6–7. [Namazova-Baranova L.S., Torshkhoeva R.M., Belyaeva I.A. Skin care of a newborn child. Guidelines. Moscow, 2016; 6–7. (in Russ)]
6. Альбанова В.И. Буллезный эпидермолиз: первый год жизни. Российский вестник перинатологии и педиатрии 2010; 55(3): 110–116. [Albanova V.I. Epidermolysis bullosa: the first year of life. Rossiyskiy Vestnik Perinatologii i Pediatrii (Russian Bulletin of Perinatology and Pediatrics) 2010; 55(3): 110–116. (in Russ)]
7. Макарова С.Г., Намазова-Баранова Л.С., МурашкинН.Н., ЕпишевР.В., ЧумбадзеТ.Р., ПетровскаяМ.И.и др. Коррекция нутритивного статуса в комплексной терапии детей, страдающих дистрофической формой врожденного буллезного эпидермолиза. Педиатрическая фармакология 2016; 13(6): 577–586. [Makarova S.G., Namazova-Baranova L.S., Murashkin N.N., Epishev R.V., Chumbadz T.R. et al. Of Nutritional Status in Complex Therapy for Children Suffering from Dystrophic Forms of Innate Epidermolysis Bullosa. Pediatricheskaya farmakologiya (Pediatric pharmacology) 2016; 13(6): 577–586. (in Russ)]
Паллиативная медицинская помощь детям с буллезным эпидермолизом — Про Паллиатив
Важным условием профилактики запоров будет наличие в рационе пищевых волокон в общем количестве 15–25 г. Потребление жидкости должно составлять минимум 100 мл/кг в сутки, что компенсирует потери жидкости через кожу, а также способствует нормальной работе кишечника, уменьшая вероятность запоров.
Дефицит витамина D сказывается на усвоении кальция, формировании ядер окостенения. Поэтому ребенку с БЭ необходимо назначать возрастную профилактическую дозу препаратов витамина D на постоянной основе [7]. Можно рекомендовать прием минерально-поливитаминных комплексов, в состав которых обязательно должны входить цинк, селен, витамин А.
Для профилактики анемии целесообразно назначать профилактическую дозу препарата железа на постоянной основе уже с первого месяца жизни (для детей с дистрофической формой). В случае развития железодефицитной анемии ее лечение у ребенка с БЭ занимает длительное время (не меньше 6 месяцев, так как железо усваивается плохо). Применяются солевые препараты железа в форме капель или сиропов, которые лучше принимать вместе с едой и не совмещать с приемом молочных продуктов. При невозможности принимать солевые препараты железа из-за развития побочных действий (чаще всего усиливаются запоры) можно прибегнуть к хелатным формам железа, которые не обладают такими побочными действиями.
Для лечения запоров используются препараты на основе лактулозы или макрогола. Важным условием их эффективности является вода, которую в достаточном количестве должен потреблять ребенок.
Для профилактики и своевременной диагностики рака кожи следует уделять внимание длительно (свыше 2 месяцев) незаживающим ранам, фотографировать их в динамике, а также по показаниям применять цитологическое исследование и биопсию. Ребенок с БЭ должен ежегодно консультироваться у детского онколога, начиная с 10-летнего возраста.
Таким образом, ведение ребенка с диагнозом БЭ – настолько непростая задача, что она непременно требует мультидисциплинарного подхода, регулярности плановых осмотров, профилактики по всем направлениям возможных осложнений. Только так можно повысить продолжительность и качество жизни детей с буллезным эпидермолизом.
Профессиональная компетентность неонатологов и их умение своевременно установить ранний диагноз БЭ у новорожденного значительно влияет на динамику и тяжесть этого заболевания у ребенка. Общеизвестно, что особенности строения кожи новорожденного способствуют ее легкой ранимости, а хорошее кровоснабжение клетчатки и отсутствие в ней соединительнотканных перемычек играют существенную роль в быстром развитии и распространении кожного процесса [9, 10].
Буллезный эпидермолиз является одним из наиболее часто встречающихся в периоде новорожденности генодерматозов (наследственных заболеваний кожи). Каждый месяц рождается ребенок с БЭ [4].
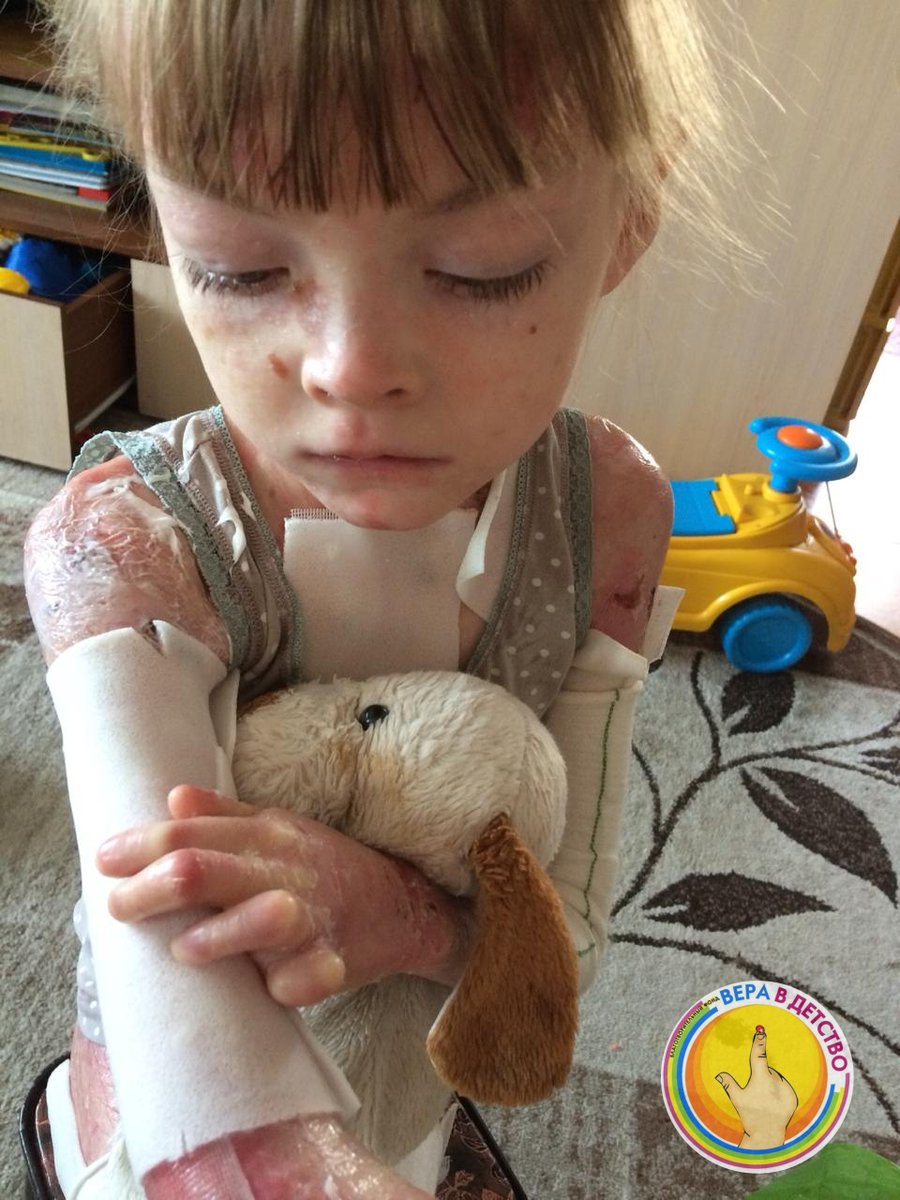
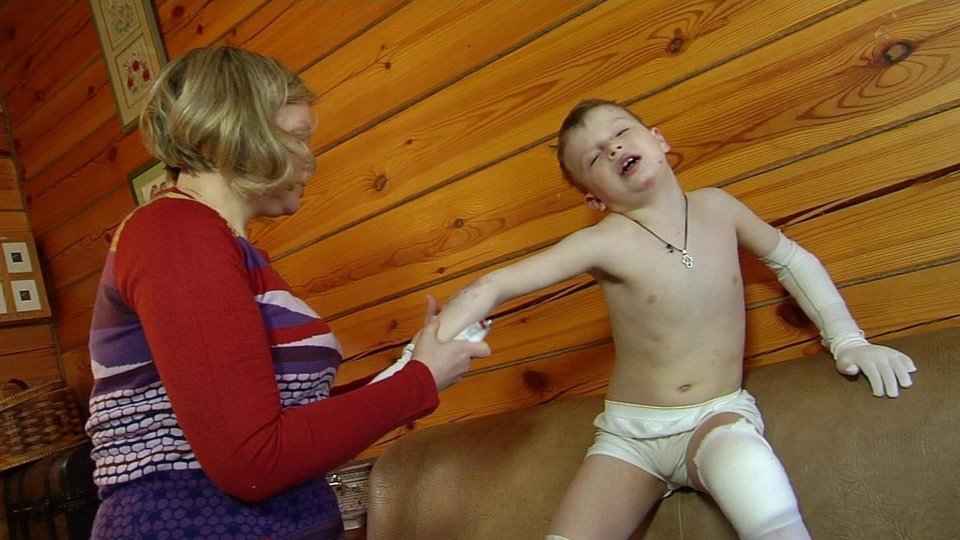
Маленькие пациенты с врожденным буллезным эпидермолизом (ВБЭ) с первых секунд жизни испытывают боль. Ее устранение играет ключевую роль с точки зрения самочувствия и качества жизни пациента [12].
В США и странах Европы существуют протоколы ведения новорожденных с БЭ, которые затрагивают все основные аспекты помощи: уход в условиях стационара, уход на дому, способы лечения и профилактики, охранительный режим, транспортировка, обучение родителей. В России таких протоколов, учитывающих неонатальные особенности БЭ, пока нет [12].
Photo: Nic co uk / Unsplash
От тактики ведения зависит дальнейшее качество жизни новорожденного и его здоровье. Врачи дерматолог и неонатолог обучают маму и других членов семьи правильному уходу за кожей ребенка. Важно в максимально ранние сроки разъяснить родителям, как правильно вести себя с ребенком-«бабочкой» в домашних условиях, на какие внекожные проявления стоит обращать внимание и когда необходимо вызывать специалиста. Уход за ребенком с БЭ часто вызывает беспокойство у родителей и медицинского персонала. Многие действия, связанные с периодом новорожденности, такие как пеленание, кормление или простая бытовая необходимость взять ребенка на руки, могут вызвать появление пузырей и эрозий [12,13].
Главными целями ухода за ребенком с ВБЭ в период новорожденности являются:
- эпителизация эрозий;
- предотвращение и лечение инфекционных осложнений;
- минимизация боли и дискомфорта;
- обеспечение оптимального питания и поддержание гармоничного физического и психомоторного развития;
- установление нормальной психологической взаимосвязи между ребенком и членами семьи.
Цели основного ухода за кожей новорожденных включают в себя:
- уменьшение и предотвращение травматических повреждений;
- защиту незрелых функций кожного барьера;
- сохранение целостности кожного покрова.
В первые дни жизни новорожденный с БЭ находится в условиях стационара, где следует соблюдать следующие правила. При необходимости постановки катетеров они должны быть зафиксированы на коже силиконовым атравматичным пластырем. Если нет возможности использовать адаптивный материал, то при удалении пластыря необходимо использовать стерильное вазелиновое масло или мазь на основе декспантенола.
При выборе подгузника необходимо, чтобы он точно соответствовал весу ребенка и не создавал дополнительного трения, а значит, пузырей, ран и, как следствие, боли.
Особое внимание уделяется выбору одежды новорожденного с ВБЭ. Швы одежды, кнопки, пуговицы могут способствовать дополнительному образованию пузырей и эрозий на коже ребенка с врожденным буллезным эпидермолизом. Одежда должна быть свободной по размеру и вывернутой швами наизнанку.
Все манипуляции необходимо проводить в перчатках, подобранных индивидуально по размеру руки того, кто выполняет манипуляцию, чтобы избежать дополнительного трения перчатки с поврежденной кожей новорожденного [4].
Обработка кожного покрова новорожденного с БЭ ни в коем случае не должна проводиться спиртосодержащим антисептиком или анилиновым красителем. Нельзя использовать перчатку как жгут или непосредственно сам жгут для фиксации выбранной области.
Купание новорожденных с БЭ имеет несколько целей:
- атравматичное снятие повязок;
- очищение кожи от корок;
- улучшение дыхательной функции кожи;
- снижение микробной колонизации [12,13].
Выписка новорожденного с БЭ из неонатального стационара в домашние условия предполагает стабильность его соматического статуса и лабораторных показателей. Родители должны быть обучены алгоритму перевязки в домашних условиях.
После выписки из стационара новорожденные с БЭ должны наблюдаться у дерматолога и педиатра, а при необходимости – и у смежных специалистов с целью мониторинга ранних осложнений заболевания. Работа клинического психолога с семьей, где есть новорожденный с БЭ, обеспечивает профессиональную психологическую поддержку родителей на этапе их адаптации к диагнозу и состоянию ребенка, а также уходу за ним.
Помочь людям оставаться родителями, даже если ребенок проживет несколько минутЧто такое перинатальная паллиативная помощь, и как она развивается в РоссииЧто знают врачи о буллезном эпидермолизе
Редкость такой неизлечимой патологии, как буллезный эпидермолиз, практически неизбежно означает ограниченный практический опыт врачей и недостаточную их осведомленность в вопросах организации современной междисциплинарной помощи пациентам с БЭ. Это приводит к поздней постановке диагноза, ошибкам в ведении пациентов, конфликтам медработников с семьей ребенка. Поиск решения данных проблем можно искать, начиная с оценки наиболее существенных пробелов во врачебных знаниях и представлениях. С данной целью в 2019 г. Фонд «БЭЛА. Дети-бабочки» и Ассоциация хосписной помощи организовали пилотную фазу анкетного опроса врачей по проблемам БЭ.
В анкетном опросе приняли добровольное участие 47 врачей различных специальностей, зарегистрированных в качестве слушателей на образовательных паллиативных форумах Ассоциации профессиональных участников хосписной помощи (АПУХП) в 2019 г. Использовался авторский опросник, разработанный совместно сотрудниками фонда «БЭЛА. Дети-бабочки» и АПУХП. Врачи опрашивались до начала образовательного симпозиума, что позволяло выяснить исходный (базовый) уровень их знаний.
Из 47 участников опроса 33 врача имели сертификаты педиатра, остальные 14 респондентов были представлены неврологами (5), детскими онкологами (2), неонатологами (2), гематологами (2), организаторами здравоохранения (2), эндоскопистами (1). Врачебный стаж респондентов составил в среднем 22,6 года, при этом врачебный стаж от 20 лет и более имели 26 участников опроса (55,3%). Личный профессиональный опыт ведения пациентов с БЭ отсутствовал у 37 респондентов (78,7%). Имели предшествующий опыт оказания медицинской помощи детям с БЭ лишь 5 респондентов из 47 (10,6%), из них 3 врача отметили, что ведут такого пациента в настоящее время.
Необходимо подчеркнуть, что при ответе на вопрос «Какие перевязочные средства вы уже назначали своим пациентам с БЭ?» 10 врачей из 47 назвали сетчатые контактные накладки на рану, а 5 врачей – губчатые накладки на кожу, столь необходимые для пациентов с БЭ.
Основные результаты опроса следует считать предварительными в связи с его небольшим объемом (табл. 1). Тем не менее опрос дает общее представление об уровне осведомленности педиатров по основным практическим аспектам ведения ребенка с БЭ.
Как следует из данных таблицы 1, наибольшее число правильных ответов было получено на вопросы No 2 (основное направление терапии БЭ), No 10 (пальчиковая гимнастика и ЛФК), а также No 14 (применение морфина внутрь перед перевязкой при выраженном болевом синдроме). Необходимо отметить, что свыше 80% респондентов адекватно оценивают показания к неинвазивным опиоидным анальгетикам в целях обезболивания при выполнении перевязок, несмотря на то, что препараты перорального морфина короткого действия в нашей стране для детей младше 18 лет еще не зарегистрированы.
Наименьшее число правильных ответов было получено на вопросы No 5 (правила забора общего анализа крови), No 9 (способы устранения неприятного запаха при уходе) и No 12 (принципы ведения запоров у ребенка с БЭ). Данный пробел в знаниях врачей обязательно нужно восполнять, так как вышеуказанные вопросы полностью относятся к компетенциям врача-педиатра, наблюдающего ребенка с БЭ. Не менее серьезный недостаток знаний врачей касается принципов местной терапии БЭ: процент правильных ответов на вопрос No 3 (ведение пузырей на коже и слизистых) составил лишь 17%, процент правильных ответов на вопрос No 4 (наружные средства, запрещенные для применения у пациентов с БЭ) – 14,9%. Правильно определяют показания к вакцинации у детей с БЭ лишь 34% респондентов (вопрос No 8). Только 36,2% врачей, принявших участие в опросе, знакомы с проблемой зуда у детей с БЭ, при том что зуд – это один из самых мучительных и трудно устранимых симптомов данного заболевания (вопрос No 13).
Имеют правильные представления о частоте смены повязок у пациента лишь 29,8% добровольных респондентов.
Таким образом, результаты опроса подтверждают крайне недостаточный уровень осведомленности врачей, работающих с детским населением, об основных практических аспектах помощи детям с БЭ. Для повышения качества помощи таким пациентам необходима разработка информационных материалов и образовательных программ.
В течение последнего года образовательные лекции экспертов фонда «БЭЛА. Дети-бабочки» включались в программы образовательных паллиативных форумов АПУХП в Иркутске, Санкт-Петербурге, Волгограде, Якутске. Фонд ведет разноплановую интенсивную работу, направленную на повышение профессиональных компетенций медицинских работников, вовлеченных в оказание помощи «детям-бабочкам». Уход за детьми с таким тяжелым, неизлечимым и ограничивающим продолжительность жизни кожным заболеванием, как буллезный эпидермолиз, – нелегкая задача, требующая особых навыков [14]. Сотрудники служб паллиативной помощи детям должны хорошо знать об этом заболевании и его симптомах, чтобы обеспечить комплексный уход в соответствии с пожеланиями ребенка и его близких.
Большинство профессионалов, работающих в сфере паллиативной помощи, редко сталкиваются с проблемами БЭ. Тем важнее будет разноплановая поддержка их компетенций профессиональными медицинскими организациями, в том числе Ассоциацией хосписной помощи.
Литература:
- Fine J. D. et al. Inherited epidermolysis bullosa: updated recommendations on diagnosis and classifi cation//J Am Acad Dermatol. – 2014.- 70 (6). – P. 1103–1126.
- Fine J. D., Hintner H. Life with Epidermolysis Bullosa (EB): Etiology, diagnosis, multidisciplinary care and therapy. Springer: Wien-New York. – 2009. – 338 p.
- Uitto J., Richard G. Progress in epidermolysis bullosa: genetic classifi cation and clinical implications//Am J Med Genet C Semin Med Genet. – 2004; 131C (1): 61–74.
- Буллезный эпидермолиз. Под ред. Дж-Д. Файна, Х. Хинтнера. Пер. с англ. Под ред. Ю. Ю. Коталевской. М.: Практика. – 2014. – С. 358.
- Трудности дифференциальной диагностики подтипов пограничного типа буллезного эпидермолиза: описание двух клинических наблюдений//Альманах клинической медицины. – 2019. – Т. 47 (1). – С. 83–93.
- Медицинские противопоказания к проведению профилактических прививок препаратами Национального календаря прививок: Методические указания МУ 3.3.1.1095–02.3.3.1. – М.: Федеральный центр госсанэпиднадзора Минздрава России, 2002. – 16 с. https://rospotrebnadzor.ru/documents/details.php?ELEMENT_ID=4716
- R. M. Kliegman, W. E. Nelson et al. Nelson textbook of pediatrics. Philadelphia, MO: Elsevier. – 2019. – 4264 p.
- А. Ю. Барановский. Диетология. Руководство. 5-е изд. – СПб: Питер. – 2019. – 1014 с. Глава 35. Белково-калорийная недостаточность.
- Зверькова Ф. А. Болезни кожи детей раннего возраста. СПб: Сотис, 1994. – C. 5–12.
- Шабалов Н. П. Неонатология, том 1. Москва: МЕДпресс-информ, 2009. – C. 162–163.
- Вулф К., Джонсон Р., Сюрмонд Д. Дерматология по Томасу Фицпатрику. – Москва: Бином, 2012. – С. 59–67.
- Denyer J. Management of the infant with epidermolysis bullosa// Infant. – 2009. – Vol 5. – P. 185–188.
- Fine J. D. Inherited epidermolysis bullosa: past, present and future// Ann NY Acad Sci. – 2010. – V. 1194 (1). – P. 213–222.
- Паллиативная помощь детям. Под ред. Э. Голдман, Р. Хейна и С. Либена. Пер. с англ. – М.: Практика, 2017. – 672 с. 27 No 3, 2019.
Авторы: Гехт Маргарита Александровна, Марычева Наталья Михайловна, Полевиченко Елена Владимировна, Туркин Алексей Олегович, Сабитова Анастасия Анатольевна, Хвостикова Елена Александровна.
Статья впервые опубликована в журнале «Pallium» # 3-2019.
НАСЛЕДСТВЕННАЯ ПУЗЫРЧАТКА (БУЛЛЕЗНЫЙ ЭПИДЕРМОЛИЗ) | Альбанова В.И.
В статье изложены клинические особенности наиболее часто встречающихся форм наследственной пузырчатки (буллезного эпидермолиза), вопросы диагностики, основные принципы лечения, возможности профилактики. Особое внимание уделено питанию, уходу, лечению осложнений и наружной терапии.
The paper outlines the clinical features of the most common types of pemphigus neonatorum (epidermolysis bullosa), diagnostic problems, the basic principles of treatment, potentialities of prevention. Particular emphasis is laid on a diet, care, treatment of complications, and topical therapy.
В.И. Альбанова, Фармацевтическое научно-производственное предприятие «Ретиноиды», главный врач Лечебно-диагностического центра «Ретиноиды», доктор мед. наук, дерматовенеролог.
V.I. Albanova, MD, dermatovenereologist, Head Physician, RETINOIDS Therapeutical-and-Diagnostic Center, RETINOIDS Pharmaceutical Research and Production Enterprise.
Наследственный
буллезный эпидермолиз — группа
пузырных наследственных кожных
заболеваний, включающая более 20
моногенных дерматозов. Так как
вопрос о самостоятельности многих
из них до настоящего времени не
решен, принято называть их формами
наследственного буллезного
эпидермолиза. Клинически общим для
всех форм является раннее начало
заболевания, чаще с рождения или
первых дней жизни, и возникновение
пузырей или эрозий на коже и
слизистых оболочках в результате
незначительной механической
травмы («механобуллезная
болезнь»). Наличие или отсутствие
рубцов после заживления дает
основание для разделения всех форм
на дистрофические и простые. С
введением в диагностику
наследственного буллезного
эпидермолиза метода электронной
микроскопии все формы стали
разделять на 3 группы: простой,
пограничный и дистрофический
буллезный эпидермолиз.
При простых формах буллезного
эпидермолиза образование пузырей
происходит в результате цитолиза
базальных эпителиоцитов, что
выявляется на электронограммах в
виде резко выраженного отека их
цитоплазмы с разрывом клеточной
оболочки. При этом неповрежденная
базальная мембрана находится в
основании пузыря. При пограничных
формах отделение эпидермиса от
дермы происходит на уровне светлой
пластинки базальной мембраны
эпидермиса из-за неполноценности
полудесмосом и крепящих
филаментов. Плотная пластинка
базальной мембраны находится в
основании пузырей. При
дистрофических формах отделение
эпидермиса от дермы происходит
ниже базальной мембраны и связано с
неполноценностью крепящих фибрилл
— структур, соединяющих базальную
мембрану с дермой.
Для проведения лечения
достаточно знать основные
клинические формы. Однако вопросы
прогнозирования и генетического
консультирования могут решаться
только на основании точного
диагноза, что в настоящее время
возможно лишь в крупных
диагностических центрах,
располагающих специалистами по
наследственным болезням кожи и
электронной микроскопии кожи
(Центральный
кожно-венерологический институт
Минздрава РФ), а также с помощью
метода иммунофлюоресцентного или
иммуногистохимического
картирования .
Кратко остановимся на основных
клинических признаках 6
существенно отличающихся друг от
друга нозологических форм
наследственной пузырчатки.
Простой генерализованный буллезный эпидермолиз
Болезнь наследуется
аутосомно-доминантно, проявляется
с рождения или первого месяца
жизни. Первые пузыри возникают на
стопах, реже на кистях, последующие
— в местах давления одежды и обуви, а
также трения (шея, поясница, локти,
колени). С возрастом количество
высыпаний уменьшается, что,
по-видимому, связано с навыками
больных избегать механических
травм. Обострение всегда
происходит в теплое время года,
хотя у маленьких детей сезонные
колебания не всегда заметны.
Пузыри и эрозии возникают через
короткое время после травмы (20 — 30
мин). Пузыри имеют полушаровидную
форму, плотную упругую покрышку,
серозное или реже кровянистое
содержимое (рис. 1). Вокруг пузыря
возникает кольцо гиперемии.
Субъективно появление пузыря
сопровождается жжением, болью,
особенно интенсивной в первые часы
после его образования. Опорожнение
пузырей приносит облегчение
больному и препятствует
дальнейшему увеличению их
размеров. Заживление происходит
быстро (2 — 3 дня), при этом покрышка
пузыря подсыхает и отслаивается.
Если покрышку пузыря срезать, то
образовавшаяся эрозия покрывается
корочкой и заживление несколько
затягивается. Период заживления
сопровождается зудом. После
заживления могут наблюдаться
легкое шелушение и пигментация.
Характерно повторное
возникновение пузырей на одном и
том же месте.
Рис. 1. Больная Н.,19 лет. Простой генерализованный буллезный эпидермолиз. Свежий пузырь полушаровидной формы с серозным содержимым.
У всех больных отмечается гипергидроз ладоней и подошв. Постепенно на местах давления и трения на подошвах формируется очаговый гиперкератоз. Изменения ногтевых пластинок (желтовато-серая окраска, утолщение, искривление) наблюдаются у всех взрослых больных. Микозы стоп нередко присоединяются к основному заболеванию.
Рис. 2. Больной К., 8 лет. Простой герпетиформный буллезный эпидермолиз. Типичные очаги округлой формы с пигментацией в центре и пузырями и корочками по периферии.
У детей младшего возраста иногда
появляются пузыри на слизистой
оболочке полости рта, однако из-за
быстрого заживления эрозий они
часто остаются незамеченными, а при
инфицировании диагностируется
афтозный стоматит.
Простой локализованный буллезный эпидермолиз
Заболевание характеризуется
теми же признаками, что и ранее
описанная форма, но высыпания
располагаются исключительно на
кистях и стопах. В литературе
описаны случаи, когда первые
признаки болезни возникали
значительно позже, чем обычно, — у
взрослых во время военной службы
или сельскохозяйственных работ при
повышении нагрузки на стопы. У
отдельных больных заболевание
начинается с распространенных
высыпаний, которые постепенно
переходят в локализованные, что
свидетельствует о генетической
общности генерализованного и
локализованного простого
буллезного эпидермолиза.
Простой герпетиформный буллезный эпидермолиз
Наследуется
аутосомно-доминантно, однако часто
встречаются спорадические случаи.
Заболевание начинается с рождения
или первой недели жизни. Первые
высыпания обычно располагаются на
кистях и стопах, быстро происходит
их распространение, что
сопровождается нарушением общего
состояния ребенка (потеря аппетита,
беспокойство, нарушение сна,
нередко подъем температуры). Пузыри
быстро эрозируются, и участки,
лишенные эпителия, занимают иногда
большую часть кожного покрова.
Развивается вторичное
инфицирование эрозий,
обезвоживание. К 3 — 6-месячному
возрасту высыпания приобретают
типичный для этой формы вид —
полушаровидные пузыри с
напряженной покрышкой и серозным,
геморрагическим или гнойным
содержимым, а также эрозии,
покрытые корочками, образуют
кольцевидные, дугообразные,
фестончатые очаги, в центре которых
расположена зона пигментации (рис.
2). Каждая отдельная эрозия заживает
быстро, заживление всего очага
растягивается на 1 — 1,5 мес.
Рис. 3. Больная С., 10 лет. Доминантный дистрофический буллезный эпидермолиз. Пузыри и эрозии на часто травмируемых участках, атрофия кожи на голенях и коленях.
Наиболее часто очаги
располагаются вокруг рта и носа, на
кистях, стопах, коленях, но может
поражаться любой участок кожи.
| Рис. 4. Больная М., 25 дней. Рецессивный дистрофический буллезный эпидермолиз. Множественные пузыри, эрозии, эритематозные пятна. | Рис. 5. Больная Л.,10 лет. Рецессивный дистрофический буллезный эпидермолиз. Эрозии, корочки, рубцовая атрофия кожи. | Рис. 7. Больной К., 6 лет. Рецессивный дистрофический буллезный эпидермолиз. Контрактуры и синдактилии стоп и кистей, обширные участки атрофии кожи. |
Прогрессирование патологического процесса может наблюдаться от нескольких месяцев до года, после чего наступает постепенное улучшение — уменьшение площади поражения кожи, более редкое возникновение пузырей и инфицирования высыпаний, нормализация общего состояния. С 2 — 3-летнего возраста у больных становится отчетливой сезонность обострений — летом высыпаний становится больше. Иногда отмечается полное разрешение высыпаний при подъеме температуры тела выше 38° С во время инфекционных заболеваний. К концу первых десяти лет жизни возникновение пузырей становится редким, а во втором прекращается.
Рис. 6. Больная Ж., 7 лет. Рецессивный дистрофический буллезный эпидермолиз. Контрактуры и синдактилии кистей.
У всех больных имеется
гиперкератоз стоп, постепенно
усиливающийся с возрастом. После
прекращения возникновения пузырей
он иногда становится единственным
симптомом заболевания.
Слизистая оболочка полости рта
поражена у большинства больных.
Эпителизация эрозий во рту
происходит очень быстро, не
оставляя следов.
Кариес, реже дефекты зубной
эмали и аномалии положения зубов
имеются у всех больных старше 2 лет.
Разнообразные изменения ногтей
наблюдаются у всех больных и
прогрессируют с возрастом. В первый
год жизни пузыри возникают иногда
под ногтевыми пластинками, которые
отслаиваются, но всегда
восстанавливаются.
Рост и развитие детей
соответствуют возрасту.
Тяжелый пограничный
буллезный эпидермолиз
Заболевание является самой
тяжелой формой наследственной
пузырчатки, приводит к смерти
ребенка в первые недели или месяцы
жизни, наследуется
аутосомно-рецессивно. Высыпания в
виде вялых, легко эрозирующихся
пузырей сразу становятся
генерализованными. Характерными
являются наличие грануляций по
краю эрозий, особенно выраженных на
лице и ногтевых валиках, тяжелое
общее состояние ребенка, резкая
анемия. Часто отслойка эпидермиса
происходит без образования пузыря.
Заживление медленное, с атрофией
кожи. В полости рта всегда имеются
эрозии. Пузыри могут
образовываться также в пищеводе,
гортани, привратнике желудка,
тонкой и прямой кишке, желчном
пузыре, уретре, почках. Больные
резко отстают в физическом
развитии. Смерть наступает в
результате асфиксии отслоившейся
покрышкой пузыря или
дерматогенного сепсиса.
Доминантный дистрофический буллезный эпидермолиз
Болезнь наследуется
аутосомно-доминантно, начинается с
рождения или первых дней жизни. В
первые месяцы поражение кожи
генерализованное, в дальнейшем
пузыри возникают обычно на одних и
тех же часто травмируемых участках:
кистях, стопах, коленях, локтях, шее.
Заживление происходит с
образованием атрофического рубца с
четкой границей, легкой
складчатостью кожи в области рубца,
пигментацией (рис.3).
Ногтевые пластинки поражены у
всех больных, и лишь в редких
случаях ногти отсутствуют, чаще они
дистрофичны. Рост и развитие детей
не нарушены. В раннем возрасте
может возникать нарушение
проходимости пищевода, что
выражается в поперхивании, рвоте
при употреблении твердой пищи,
слюнотечении, боли при глотании.
Эти явления обратимы.
С возрастом пузыри появляются
все реже, и у взрослых о наличии
болезни могут напоминать только
дистрофические изменения ногтей и
едва заметные рубцы на локтях,
коленях и лодыжках.
Рецессивный дистрофический буллезный эпидермолиз
Болезнь наследуется
аутосомно-рецессивно, протекает
тяжело, часто приводит к смерти в
раннем возрасте. Заболевание
всегда возникает с рождения или
первых часов жизни. Уже при
рождении часто эрозирована кожа
конечностей. В первые дни жизни
происходит распространение
высыпаний, причем пузыри возникают
не только в результате легких травм
кожи, давления и трения, но и
спонтанно (рис. 4). Даже крупные
эрозии заживают сравнительно
быстро (в зависимости от размеров
за 3 — 10 дней), но постоянно
появляются новые (рис. 5). Заживление
происходит с образованием
атрофических рубцов, на кистях и
стопах постепенно развиваются
контрактуры и синдактилии (рис. 6,7).
Ногтевые пластинки отсутствуют с
рождения или постепенно
утрачиваются в результате
образования подногтевых пузырей.
Рубцовая атрофия кожи волосистой
части головы проявляется диффузной
разреженностью волос и их
дистрофическими изменениями.
На слизистой оболочке полости
рта, пищевода, прямой кишки также
возникают множественные пузыри.
Процесс рубцевания во рту приводит
к ограничению подвижности языка,
атрофии его сосочков, заращению
вестибулярных складок и
микростомии, в пищеводе — к его
сужению, нарушению проходимости
пищи, в прямой кишке — к хроническим
запорам, резким болям при
дефекации.
Зубы поражены у всех больных,
преобладают кариес, дефекты зубной
эмали, аномалии расположения.
Поражение слизистой оболочки
глаз часто наблюдается в детском
возрасте, что клинически
проявляется жжением, болями при
попытке открыть глаза.
Эпителизация эрозий конъюнктивы
происходит быстро (2 — 4 дня).
Рубцевание на роговице завершается
образованием облачковидных
помутнений, существенно не
нарушающих зрение.
Общее состояние больных
характеризуется слабостью, быстрой
утомляемостью, длительными
периодами субфебрилитета.
Постоянные болезненные ощущения
приводят к ограничению подвижности
больных, замедлению психомоторного
и физического развития, социальной
дезадаптации. Отмечается
гипохромная анемия.
С возрастом способность к
заживлению эрозивноязвенных
поражений снижается, некоторые
очаги не заживают несколько
месяцев и даже лет. На таких очагах,
а также на рубцах могут
формироваться эпителиальные
опухоли, чаще плоскоклеточный рак,
резистентный к терапии.
Среди причин смерти в первый год
жизни наиболее часты асфиксия,
аспирационная пневмония,
дерматогенный сепсис, в возрасте
старше 30 лет — злокачественные
опухоли кожи.
Лечение наследственной
пузырчатки
Разрабатывая тактику лечения,
следует учитывать, что период
активности наследственных
болезней растягивается на долгие
годы и вряд ли стоит рекомендовать
столь же длительный прием даже
эффективных препаратов. Активная
терапия применяется во время
обострения и возникновения
осложнений короткими курсами, а
затем сменяется более длительным
назначением общеукрепляющих и
симптоматических средств. Особое
внимание уделяется вопросам ухода
и питания.
Основными задачами лечения
являются предупреждение появления
пузырей на коже и слизистых
оболочках, ускорение заживления
имеющихся высыпаний, профилактика
и лечение вторичного
инфицирования, предупреждение и
лечение тяжелых осложнений,
связанных с рубцеванием.
Патогенетическая терапия
Известно, что в коже больных
рецессивным дистрофическим
буллезным эпидермолизом
вырабатывается избыточное
количество структурно измененной
коллагеназы. В связи с этим
патогенетически обосновано
применение препаратов,
ингибирующих выработку или
активность коллагеназы — дифенина
(фенитоина), эритромицина, больших
доз витамина Е и ретиноидов.
Дифенин (применяемый в
психиатрии для лечения эпилепсии)
выпускают в таблетках по 0,1,
назначают внутрь дважды в день из
расчета 3,5 мг/кг массы тела в сутки у
взрослых и 8 мг/кг у детей. В течение
первых 3 дней назначают 1/3 суточной
дозы, с 4-го по 6-й день 2/3, с 7-го дня —
полную дозу. При необходимости
отмены препарата снижение его дозы
производят в обратном порядке.
Положительный эффект в виде
уменьшения количества пузырей,
ускорения эпителизации эрозий,
повышения резистентности кожи к
травматическим воздействиям
отмечают через 3 — 4 нед. При лечении
дифенином возможны головокружение,
возбуждение, тошнота, рвота, тремор,
лимфаденопатия, гиперплазия десен.
Эритромицин назначают внутрь
в обычной возрастной дозе в течение
10 — 14 дней. Учитывая
бактериостатическое действие
антибиотика, его лучше назначать,
когда множественные пузырные
высыпания на коже сопровождаются
вторичным инфицированием эрозий.
Токоферола ацетат (витамин Е)
выпускают в капсулах по 50 и 100 мг и в
масляном растворе 5%, 10% и 30% ( по 50, 100
и 300 мг/мл). Препарат при буллезном
эпидермолизе дает положительный
эффект только в дозах, превышающих
1500 мг в сутки. Детям назначают 1/2 — 1/3
этой дозы (то есть 500 — 1000 мг в сутки).
Курс лечения составляет 20 — 40 дней.
Препарат назначают равными частями
утром и вечером во время или сразу
после еды. Детям старшего возраста,
не имеющим нарушений глотания, и
взрослым витамин Е назначают в
капсулах, в остальных случаях — в
каплях. Побочных явлений при
применении таких высоких доз нами
не отмечено и в литературе не
описано.
Из группы ретиноидов применяются тигазон и ретинола пальмитат или ретинола ацетат. Помимо
антиколлагеназного действия, они
обладают способностью ускорять
эпителизацию. В связи с этим их
лучше назначать больным со
значительной площадью поражения,
но без вторичного инфицирования.
Тигазон выпускают в капсулах по 10 и
25 мг, назначают в суточной дозе 1
мг/кг массы тела (равными частями 3
раза в день во время еды). При
хорошем эффекте (обычно на 3 — 7-й
день) через 2 — 3 нед суточную дозу
снижают до 0,3 — 0,5 мг/кг. Длительное
применение тигазона не
рекомендуется, поскольку препарат
обладает множеством побочных
действий: хейлит, сухость кожи и
слизистых оболочек, выпадение
волос, биохимические нарушения, у
детей — замедление роста. Тигазон
эмбриотоксичен и тератогенен,
противопоказан беременным и
планирующим беременность женщинам.
Перед началом лечения необходимо
убедиться, что в клиническом и
биохимическом анализах крови нет
отклонений.
Ретинола пальмитат выпускают в
капсулах по 100000 МЕ и масляном
растворе по 100000 МЕ/мл, ретинола
ацетат — в капсулах по 3300, 5000 и 33000МЕ
и масляном растворе по 100000 и 250000
МЕ/мл. Оба препарата применяются в
суточной дозе 5000 МЕ/кг массы тела
(равными частями утром и вечером
после еды). По сравнению с тигазоном
препараты менее токсичны, но их
положительное действие
проявляется в более поздние сроки
(на 7 — 10-й день). Курс лечения
составляет 1,5 — 2 мес.
Препараты с антиколлагеназной
активностью не всегда эффективны.
Большинство из них не
рекомендуется применять длительно,
обычно проводят 2 — 3 курса лечения в
год. Основой лечения остается
симптоматическая терапия.
Симптоматическа терапия
Общая терапия включает
применение антибиотиков широкого
спектра действия при вторичном
инфицировании высыпаний (их
назначают только при нарушении
общего состояния больного),
антигистаминные и седативные
препараты при выраженном зуде,
анаболические и общеукрепляющие
средства и ферменты при отставании
в физическом развитии. При анемии
лучший эффект дает прямое
переливание крови, возможно
переливание эритроцитной массы,
плазмы, альбумина, обязательно
назначение соответствующей диеты.
В комплексной терапии
необходимо назначение
поливитаминных препаратов.
Предпочтение отдают комплексам,
содержащим микроэлементы.
При поражении слизистой
оболочки полости рта после каждого
приема пищи рекомендуется
прополоскать рот отварами ромашки,
шалфея, календулы, зверобоя,
дубовой коры, корневища змеевика.
После полоскания хорошо применять
облепиховое масло, мазь с ретинола
пальмитатом, каротолин, солкосерил,
сок каланхое в виде аппликаций.
Маленьким детям дают облепиховое
масло в каплях внутрь.
При спазмах пищевода с
нарушением проходимости пищи
рекомендуются постельный режим,
сухое тепло на область груди,
спазмолитические средства,
электрофорез с пелоидином.
Уход за кожей и наружное лечение
При запорах не следует применять
слабительные средства, вызывающие
химическое или механическое
раздражение рецепторов кишечника,
лучший эффект достигается диетой и
введением в прямую кишку теплого
растительного или вазелинового
масла в виде микроклизмы.
Применение кортикостероидных
препаратов как внутрь, так и
наружно не рекомендуется. Не
препятствуя образованию пузырей и
не способствуя заживлению, они
усиливают формирование
атрофических рубцов.
Профилактические прививки
противопоказаны только в период
нарушения общего состояния
ребенка.
С первых дней жизни больного
ребенка нужно стремиться свести к
минимуму травмирующие кожу
факторы. Пеленки должны быть
мягкими, без швов. При пеленании
обращают внимание на то, чтобы
ножки ребенка не соприкасались
друг с другом. Все нижнее белье
надевается швами наружу.
На одежде не должно быть резинок
и тесемок, стесняющих движения. По
этой же причине нежелательно
пользоваться памперсами.
На руки ребенка берут очень
осторожно, только одетым или
завернутым в пеленку, поддерживая
снизу, избегая растягивания кожи.
Когда ребенок начинает ходить, в
его одежду (на локти и колени)
желательно подшить несколько слоев
мягкой ткани. Дома рекомендуется
носить свободную одежду и не
надевать обуви. Обувь для прогулок
должна быть свободной и легко
надеваться.
Все лица, причастные к уходу за
ребенком и наблюдению за
состоянием его здоровья, должны
быть предупреждены об особенностях
болезни и недопустимости даже
незначительного травмирования
кожи и слизистых оболочек
(например, при осмотре педиатром,
отоларингологом или стоматологом).
Пузыри легче образуются на
сухой, атрофичной коже со сниженным
салоотделением. В связи с этим кожу
нужно ежедневно искусственно
ожиривать и гидратировать, чтобы
она приобретала мягкость и
эластичность и была более
резистентна к механическим
воздействиям. С этой целью
применяются кремы и мази на
водно-эмульсионной основе (мазь с
ретинола пальмитатом, мазь радевит,
5% мазь с мочевиной) или
растительное масло (кроме
подсолнечного). Используемый крем
не должен быть густым.
Пузыри, образующиеся на коже,
ежедневно опорожняют, прокалывая с
двух сторон толстой инъекционой
иглой и удаляя жидкость либо
надавливанием, либо отсасыванием с
помощью шприца.
Следует помнить, что собственный
эпидермис является самым лучшим
покрытием раны и более всего
способствует ее заживлению. Однако
в большинстве случаев повторная
механическая травма приводит к
разрыву отслоившегося эпидермиса,
обнажается эрозивная поверхность.
На эрозии лучше всего сразу же
наложить коллагеновое губчатое
покрытие (коласпон, альгикол,
дигиспон и др.). Если покрытий нет
под рукой, необходимо обработать
ранку антисептиком во избежании
инфицирования и наложить
стерильную повязку с
ранозаживляющими средствами (мази
с ретинола пальмитатом, радевит,
солкосерил, актовегин, бепантен,
аэрозоль пантенол).
Несмотря на предосторожности,
большинство эрозий инфицируется,
превращается в язвы. На
инфицированные участки
накладывают мази с антибиотиками
или другими антимикробными
средствами (дермазин, левосин,
левомеколь, фастин). Удобны
аэрозольные наружные средства: олазоль,
гипозоль, легразоль, левовинизоль и
др. Для заживления язв
используются коллагеновые
губчатые покрытия, содержащие
ферменты.
При обширном поражении хороший
эффект оказывает общее
УФ-облучение субэритемными дозами.
Магнитотерапию назначают на
отдельные длительно незаживающие
язвы.
Ванны с травами хорошо
стимулируют заживление, улучшают
самочувствие и являются для
больных любого возраста самой
приятной процедурой. Для ванн
используются любые растительные
отвары с противовоспалительным и
вяжущим действием. При выраженном
зуде добавляют мяту или валериану.
После ванны обязательно смазывание
кожи питательным кремом.
У многих больных после
заживления образуются милиумы,
особенно часто на тыльной стороне
кистей. Эти высыпания со временем
разрешаются сами и не требуют
лечения.
Взрослые больные должны быть
предупреждены о возможном
появлении на рубцах и язвах
необычных высыпаний, что требует
незамедлительного обращения к
дерматологу. Эпителиальные опухоли
при буллезном эпидермолизе могут
быть резистентны к лечению, оно
должно быть начато как можно
раньше.
Питание
Во все периоды жизни больного питание должно компенсировать потери белка, солей и воды, связанные с образованием пузырей. Грудное вскармливание предпочтительнее, с введением докорма и прикорма количество вводимого с пищей белка должно быть увеличено на 20%. Фруктовые соки и пюре не должны быть кислыми. В утренние часы полезно давать ребенку немного нерафинированного растительного масла, которое восполняет недостаток полиненасыщенных жирных кислот и облегчает дефекацию. В рацион вводят продукты, богатые грубой растительной клетчаткой (капуста, кабачки, свекла, сухофрукты). Запрещается пища, травмирующая слизистую оболочку (карамель, сухари, сушки, вафли и т. д). Взрослых больных предостерегают от употребления спиртных напитков и острых блюд, провоцирующих образование пузырей в пищеводе.
При спазмах и сужении пищевода применяют механически и термически щадящую диету, увеличивая количество приемов пищи.
Проф. С. И. Воздвиженским (МНИИ
педиатрии и детской хирургии)
разработан метод хирургического
лечения контрактур и синдактилий
кистей у детей с буллезным
эпидермолизом. Однако существует
опасность рецидивов, в связи с чем
необходим длительный период
противорецидивной терапии.
Больные дети могут получить
инвалидность, взрослые
трудоспособны или являются
инвалидами I — III группы в
зависимости от формы, тяжести
заболевания и осложнений.
В связи с трудностями лечения
особое значение имеют методы
профилактики. Генетическое
консультирование возможно после
установления точного диагноза.
Риск развития заболевания при
доминантных формах — 50% для каждого
ребенка. Если у здоровых родителей
родился ребенок, больной
рецессивной формой буллезного
эпидермолиза, то для каждого
следующего ребенка риск развития
заболевания составляет 25%. Если
больной с рецессивной формой хочет
иметь ребенка, то для него риск
развития заболевания ничтожно мал.
Риск становится большим (50%) в том
случае, если второй из родителей
является носителем идентичного
рецессивного гена буллезного
эпидермолиза. Риск существенно
увеличивается при
кровнородственных браках.
При пограничном и рецессивном
дистрофическом буллезном
эпидермолизе возможно проведение
пренатальной диагностики методом
биопсии кожи плода в 16 — 18 нед
беременности с последующим
электронно-микроскопическим
исследованием. Такое исследование
может быть проведено в Центре
охраны здоровья матери и ребенка
(Москва) и в ряде зарубежных стран.
Если у плода имеется заболевание,
производят прерывание
беременности.
Литература:
1. Bruckner, Tuderman L. Epidermolysis bullosa
hereditary. Hautarzt 1995;46:61-72.
2. Альбанова В.И. Клиническая
характеристика доминантного
дистрофического буллезного
эпидермолиза. Вестник дерматол. —
1994;1:48-52.
3. Альбанова В.И. Буллезный
эпидермолиз. В книге «Моногенные
дерматозы». — Йошкар-Ола. — 1993. С.
104-26.
4. Суворова К.Н., Альбанова В.И.
Наследственный буллезный
эпидермолиз. В книге «Детская
дерматовенерология. — Казань. 1996. С.
69-80.
Буллезный эпидермолиз — NORD (Национальная организация по редким заболеваниям)
ТЕКСТОВЫЕ КНИГИ
Fine JD, Hintner H., eds. Жизнь с буллезным эпидермолизом (БЭ): этиология, диагностика, многопрофильная помощь и терапия, Нью-Йорк: Springer Wien; 2009.
Fine JD, Bauer E, McGuire J, Moshell A, eds. Буллезный эпидермолиз: клинические, эпидемиологические и лабораторные достижения и результаты Национального реестра буллезного эпидермолиза 1-е издание The Johns Hopkins University Press; 1999 г.
Weston WL, Lane AT, Morelli JG, ред. Цветной учебник детской дерматологии, 4-е издание Mosby / Elsevier, 2007: 348-354.
ОБЗОР СТАТЬИ
Мюррелл Д.Ф. (ред.) (2010): Буллезный эпидермолиз: Часть I — Патогенез и клинические особенности. Дерматологические клиники, Том 28/1. Филадельфия: W.B. Saunders Elsevier.
Мюррелл Д.Ф. (ред.) (2010): Буллезный эпидермолиз: Часть II — Диагностика и лечение. Дерматологические клиники, Том 28/2. Филадельфия: W.B. Saunders Elsevier.
Fine JD, Mellerio JE. Внекожные проявления и осложнения наследственного буллезного эпидермолиза: часть I. Ткани, ассоциированные с эпителием.
J Am Acad Dermatol. 2009 сентябрь; 61 (3): 367-84.
Fine JD, Mellerio JE. Внекожные проявления и осложнения наследственного буллезного эпидермолиза: часть II. Другие органы. J Am Acad Dermatol. 2009 сентябрь; 61 (3): 387-402.
Pfendne EG, Bruckner A, Conget P, Mellerio J, Palisson F, Lucky AW. Фундаментальная наука о буллезном эпидермолизе, его диагностическая и молекулярная характеристика: материалы II Международного симпозиума по буллезному эпидермолизу.Сантьяго, Чили, 2005 Международный журнал дерматологии. 2007; 46 (8): 781-794.
Азизхан Р.Г., Дениер Дж.Э., Меллерио Дж.Э., Гонсалес Р., Бачигалупо М., Кантор А., Пассалаква Г., Палиссон Ф., Лаки А.В. Хирургическое лечение буллезного эпидермолиза: материалы II Международного симпозиума по буллезному эпидермолизу, Сантьяго, Чили, 2005 г. Международный журнал дерматологии. 2007: 46 (8): 801-808.
Mellerio JE, Weiner M, Denyer JE, Pillay EI, Lucky AW, Bruckner A, Palisson F. Медицинское лечение буллезного эпидермолиза: Материалы II Международного симпозиума по буллезному эпидермолизу, Сантьяго, Чили, 2005 Международный журнал дерматологии 2007; 46 (8): 795-800.
Lucky AW, Pfendner E, Pillay E, Paskel J, Weiner M, Palisson F. Психосоциальные аспекты буллезного эпидермолиза: Материалы II Международного симпозиума по буллезному эпидермолизу, Сантьяго, Чили, 2005 Международный журнал дерматологии. 2007; 46 (8): 809-814.
ИНТЕРНЕТ
Debra International. Уход за больным БЭ. Доступно по адресу: http://www.debra-international.org/patients/caring-for-someone-with-eb.html По состоянию на 30 мая 2013 г.
Маринкович М.П.Буллезный эпидермолиз Обновлено: 10 августа 2012 г. Medscape. Доступно по адресу: http://emedicine.medscape.com/article/1062939-overview По состоянию на 30 мая 2013 г.
Пфенднер Э., Брукнер А. Простой буллезный эпидермолиз. Последнее обновление 1 сентября 2011 г. В: GeneReviews at GeneTests: Medical Genetics Information Resource (онлайн-база данных). Авторские права, Вашингтонский университет, Сиэтл. 1997-2013. Доступно на http://www.genetests.org. По состоянию на 30 мая 2013 г.
Pfendner E, Lucky AW. Дистрофический буллезный эпидермолиз.Последнее обновление 4 ноября 2010 г. В: GeneReviews at GeneTests: Medical Genetics Information Resource (онлайн-база данных). Авторские права, Вашингтонский университет, Сиэтл. 1997-2013. Доступно на http://www.genetests.org. По состоянию на 30 мая 2013 г.
Pfendner E, Lucky AW. Буллезный узловой эпидермолиз. Размещено: 2 февраля 2008 г. В: Обзоры генов на сайте GeneTests: информационный ресурс по медицинской генетике (онлайн-база данных). Авторские права, Вашингтонский университет, Сиэтл. 1997-2013. Доступно по адресу http: //www.genetests.орг. По состоянию на 30 мая 2013 г.
Pfendner E, Lucky AW. Буллезный эпидермолиз с атрезией привратника. Последнее обновление: 14 февраля 2013 г. В: GeneReviews at GeneTests: Medical Genetics Information Resource (онлайн-база данных). Авторские права, Вашингтонский университет, Сиэтл. 1997-2013. Доступно по адресу http://www.genetests.org По состоянию на 30 мая 2013 г.
Буллезный эпидермолиз — NHS
Буллезный эпидермолиз (БЭ) — это название группы редких наследственных кожных заболеваний, при которых кожа становится очень хрупкой.Любая травма или трение кожи могут вызвать болезненные волдыри.
Информация:Консультации по коронавирусу
Получите консультацию по коронавирусу и буллезному эпидермолизу (БЭ) от DEBRA
Симптомы буллезного эпидермолиза
Основные симптомы всех типов БЭ включают:
- кожа, которая легко покрывается волдырями
- волдыри во рту
- волдыри на руках и подошвах ног
- кожа с рубцами, иногда с небольшими белыми пятна, называемые милиами
- утолщение кожи и ногтей
Кредит:
Типы буллезного эпидермолиза
Три основных типа БЭ:
- простой буллезный эпидермолиз (EBS) — наиболее распространенный тип, который имеет тенденцию быть более легким с низким риском серьезных осложнений
- дистрофический буллезный эпидермолиз (DEB) — от легкой до тяжелой
- Буллезный узловой эпидермолиз (JEB) — самый редкий и наиболее тяжелый тип
Тип отражает, где на теле появляются волдыри и какой слой кожи поражен .
Существует также множество вариантов этих трех основных типов БЭ, каждый из которых имеет несколько разные симптомы.
Подробнее о симптомах различных типов буллезного эпидермолиза.
Диагностика EB
EB обычно диагностируется у младенцев и детей вашей неонатальной бригадой, поскольку симптомы часто очевидны с рождения. Но некоторые более легкие типы БЭ могут быть диагностированы только в зрелом возрасте.
Если есть подозрение, что у вашего ребенка такое заболевание, его направят к кожному специалисту (дерматологу).
Специалист проведет анализы для определения типа БЭ и поможет составить план лечения. Они могут взять небольшой образец кожи (биопсия) для отправки на тестирование.
Пренатальное тестирование
В некоторых случаях можно проверить еще не родившегося ребенка на БЭ примерно на 11 неделе беременности.
Это может быть предложено, если известно, что вы или ваш партнер являетесь носителем дефектного гена, связанного с БЭ, и есть риск рождения ребенка с тяжелым типом БЭ.
Если тест подтвердит, что у вашего ребенка БЭ, вам будут предложены консультации и рекомендации, которые помогут вам принять обоснованное решение о том, как вы хотите продолжить беременность.
Пренатальные тесты включают амниоцентез и забор проб ворсин хориона.
Причины буллезного эпидермолиза
EB вызывается дефектным геном (генная мутация), который делает кожу более хрупкой.
Обычно ребенок с EB унаследует дефектный ген от родителя, у которого также есть EB.
Также возможно, что ребенок с EB унаследовал дефектный ген от обоих родителей, которые являются просто «носителями», но сами не имеют EB.
Лечение буллезного эпидермолиза
В настоящее время лекарства от БЭ не существует, поэтому лечение направлено на облегчение симптомов и предотвращение развития осложнений, таких как инфекция.
Группа медицинских специалистов поможет вам решить, какое лечение лучше всего подходит для вашего ребенка, и посоветует, как жить с этим заболеванием.
Вы можете лечить БЭ в домашних условиях:
- вскрывать волдыри стерильной иглой
- накладывать защитные повязки
- избегать вещей, ухудшающих состояние
Лекарства можно использовать для лечения инфекции или уменьшения боли. Операция может потребоваться, если БЭ вызывает сужение пищевода или проблемы с руками.
Подробнее о лечении буллезного эпидермолиза.
Приобретенный буллезный эпидермолиз
Приобретенный буллезный эпидермолиз (ББЭ) — приобретенная форма БЭ с аналогичными симптомами.
Как и EB, EBA легко вызывает образование волдырей. Это также может повлиять на ротовую полость, горло и пищеварительный тракт.
Но EBA не передается по наследству, и симптомы обычно не проявляются до более позднего возраста.
Это аутоиммунное заболевание, которое означает, что ваша иммунная система начинает атаковать здоровые ткани тела. Точно не известно, что вызывает это.
EBA — очень редкое заболевание, которое чаще встречается у людей старше 40 лет.
Благотворительные организации и группы поддержки
Если вашему ребенку поставили диагноз БЭ, это может быть пугающим и подавляющим опытом.Вероятно, вы захотите узнать как можно больше о состоянии и доступных методах лечения.
DEBRA — это национальная благотворительная организация, которая предоставляет помощь, советы и поддержку людям в Великобритании, живущим с EB.
DEBRA International — это всемирная сеть национальных групп, работающих от имени людей, пострадавших от БЭ.
Поддержка лиц, осуществляющих уход
При уходе за ребенком со сложным и тяжелым заболеванием, таким как БЭ, важно не пренебрегать своим здоровьем и благополучием.
Прочтите о перерывах в уходе и временном уходе и получите советы по уходу за ребенком-инвалидом.
Информация о вас
Если у вас или вашего ребенка есть БЭ, ваша клиническая бригада передаст информацию о вас или вашем ребенке в Национальную службу регистрации врожденных аномалий и редких заболеваний (NCARDRS).
Это помогает ученым искать более эффективные способы профилактики и лечения этого состояния. Вы можете отказаться от регистрации в любое время.
Подробнее о реестре
Последняя проверка страницы: 19 марта 2018 г.
Срок следующей проверки: 19 марта 2021 г.
Буллезный эпидермолиз | DermNet NZ
Автор: Ванесса Нган, штатный писатель, 2003 г. Обновлено Джейн Уиддоусон, специалистом по клинической медсестре, и др., DEBRA, Новая Зеландия, февраль 2016 г.
Что такое буллезный эпидермолиз?
Буллезный эпидермолиз (БЭ) — это группа наследственных заболеваний, которые характеризуются образованием пузырей на коже и слизистых оболочках.Они могут возникать на любом участке тела, но чаще всего появляются в местах трения и незначительных травм, таких как ступни и руки. В некоторых подтипах волдыри могут также возникать на внутренних органах, таких как пищевод, желудок и дыхательные пути, без видимого трения.
Буллезный эпидермолиз
EB следует отличать от обычных волдырей от трения и от приобретенного буллезного эпидермолиза (EBA), который представляет собой пузырчатое аутоиммунное заболевание, которое не передается по наследству и часто не развивается до взрослой жизни.
Заболевания БЭ возникают в результате генетических дефектов молекул кожи, связанных с адгезией. Потеря адгезии приводит к образованию пузырей. Существует четыре основных типа БЭ, основанных на различных участках образования волдырей в структуре кожи:
Внутри каждого из этих типов БЭ существуют различные подтипы. Каждый тип БЭ имеет разную степень тяжести, от легкой до тяжелой. Международный консенсус в отношении диагностики и классификации БЭ привел к обновленным рекомендациям (2014 г.), основанным на новых клинических и молекулярных данных.
Кто заболевает буллезным эпидермолизом?
EB — это наследственное заболевание, что означает, что вы унаследовали один или два гена EB. При аутосомно-доминантном БЭ для экспрессии болезни необходим только один аномальный ген. Это означает, что только один родитель должен нести ген EB. С другой стороны, аутосомно-рецессивный наследственный EB требует, чтобы у вас было два гена EB (по одному от каждого родителя), чтобы иметь заболевание. Если у человека есть один рецессивный ген EB в паре с нормальным геном, его называют носителем, и он не болен.
БЭ обычно возникает при рождении или вскоре после него. В равной степени страдают самцы и самки. Иногда БЭ может быть достаточно легкой при рождении, чтобы не проявляться, и обнаруживается только тогда, когда ребенок подрастет или не достигнет совершеннолетия.
Каковы клинические признаки буллезного эпидермолиза?
Простой буллезный эпидермолиз (EBS)
Подробнее см. Простой буллезный эпидермолиз.
| Подтипы EBS | Характеристики |
|---|---|
| Локализованный EBS Ранее известный как Weber-Cockayne |
|
| Generalized EBS Ранее известный как Koebner |
|
| Общая тяжелая форма EBS Ранее известная как Даулинг Меара |
|
Буллезный соединительный эпидермолиз (JEB)
Подробнее см. Буллезный соединительный эпидермолиз.
| Подтипы JEB | Характеристики |
|---|---|
| Общая тяжелая форма JEB Ранее известный как Herlitz |
|
| Обобщенный промежуточный JEB Ранее известный как Non-Herlitz |
|
Буллезный дистрофический эпидермолиз (DEB)
Подробнее см. Буллезный дистрофический эпидермолиз.
| Подтипы DEB | Характеристики |
|---|---|
| Доминантный обобщенный DEB |
|
| Генерализованный тяжелый рецессивный (R) DEB Ранее известный как Hallopeau-Siemens; а также; Обобщенный промежуточный RDEB (ранее Non-Hallopeau-Siemens) |
|
Синдром Киндлера
| Синдром Киндлера | Характеристики |
|---|---|
| Синдром Киндлера |
|
Есть много других подтипов EB.На проявление и тяжесть БЭ влияют специфические генетические изменения, которые иногда бывает трудно классифицировать.
Генетическое консультирование и пренатальное тестирование на БЭ доступны в некоторых центрах, и их следует рассмотреть.
Направление за психологической поддержкой также следует рассматривать для детей с БЭ и их семей, особенно для более тяжелых подтипов.
Как диагностируется буллезный эпидермолиз?
В доминирующих подтипах БЭ, для которых известно информативное генеалогическое древо, часто приемлемо, чтобы клинический диагноз (на основе имеющихся признаков) был поставлен специалистом-дерматологом.
Диагностические тесты также доступны в некоторых странах и включают биопсию кожи недавно образованного волдыря, которая подвергается иммунофлуоресцентному картированию антигенов (IFM) и просвечивающей электронной микроскопии (EM). Мутационный анализ (анализ крови на гены), хотя в настоящее время не считается диагностическим тестом первой линии, также доступен в некоторых странах.
Требуется направление к дерматологу.
Как оценивается степень тяжести буллезного эпидермолиза?
Тяжесть EB можно оценить с помощью следующих систем оценки:
- Оценка Birmingham EB
- EBDASI: Индекс активности БЭ и рубцов
- iSCOREB: инструмент для оценки клинических результатов исследования EB
- QOLEB: оценка качества жизни в EB
Лечение буллезного эпидермолиза
Общие
Лекарства от БЭ не существует.Однако значительные исследования, включая генную терапию и клеточную терапию, продолжаются с целью улучшения качества жизни.
В большинстве случаев лечение симптоматическое. Основная цель — защитить кожу и остановить образование волдырей, ускорить заживление и предотвратить осложнения.
Поскольку БЭ может поражать очень много различных частей тела, обычно требуется группа медицинских специалистов для оказания общей помощи. При необходимости врач может назначить лечение пероральными и местными лекарствами для облегчения заживления или предотвращения осложнений.
Ниже приведены некоторые общие меры по уходу за пациентом с БЭ.
- Избегайте действий, вызывающих трение кожи. Это включает в себя обращение с младенцами и детьми — альтернативным методам обращения с ними легко научиться у квалифицированного специалиста в области здравоохранения.
- Поддерживайте прохладу и избегайте перегрева
- Используйте набивку из пеноматериала или овчину, чтобы уменьшить трение о предметы мебели, такие как кровати, стулья и детские автокресла
- Выбирайте легкую одежду (включая подгузники) и обувь, не имеющую раздражающих швов или деталей (например, молнии и тугую резинку).
- Прокалывание, дренирование и обработка волдырей для ускорения заживления (это должны делать только люди, прошедшие обучение по уходу за ранами)
- Многие традиционные липкие ленты и повязки могут быть неподходящими для людей с БЭ, особенно с более тяжелыми формами (например, RDEB), поскольку их удаление может вызвать дополнительную травму кожи. Рекомендуется использование современных средств ухода за ранами, таких как силиконовые ленты и повязки с низким сцеплением. Однако находчивость в использовании легкодоступных предметов, таких как нанесение дополнительной смазки (например, вазелин / парафиновое масло) на некоторые традиционные повязки для ран, может оказаться полезной.
- Когда БЭ поражает другие части тела, применяются различные методы ухода и лечения. Например, мягкая диета при поражении пищевода или использование смягчителей стула при запоре, или если у пациента есть анальные волдыри.
- Руководства по клинической практике доступны на веб-сайте DEBRA * International, в том числе руководства по уходу за полостью рта, уходу за ранами, лечению боли, лечению рака.
- Дополнительная поддержка и информация как от специалистов здравоохранения, так и от лиц, пострадавших от БЭ:
- DEBRA (Исследовательская ассоциация буллезного дистрофического эпидермолиза) имеет более 50 групп по всему миру.
- Eb-Clinet — это клиническая сеть центров EB и экспертов по всему миру.
Буллезный эпидермолиз, худшее заболевание, о котором вы никогда не слышали
Характерная чертаЭлиза Бэрд родилась с редким генетическим заболеванием, из-за которого ее кожа была хрупкой, как крылья бабочки. Она была одной из «Детей бабочек».
Элиза Бэрд, у которой было очень редкое заболевание кожи — буллезный эпидермолиз.Предупреждение. Эта статья содержит графические изображения.
Все отсканированные изображения вернулись в норму. По общему мнению, беременность протекала гладко.
Но когда в 2000 году Симона Бэрд родила дочь, вскоре стало ясно, что что-то не так.
«Вся ее ступня была сырой, на ней не было никакой кожи, а колено — прямо по голени», — сказала г-жа Бэрд.
«У нее было немного кожи на тыльной стороне ладони, и ее язык также был затронут.
«Она была … идеальным ребенком во всех отношениях, но без кожи. Вошел педиатр и сказал: «Если бы я не знал ничего лучше, я бы сказал, что вы облили ее кипятком».
Элизу срочно отправили в Королевскую детскую больницу, а молодая мать осталась в частной больнице, наблюдая, как другие женщины связываются со своими младенцами.
Через две недели у новорожденного был диагностирован рецессивный дистрофический буллезный эпидермолиз (БЭ), тяжелая форма редкого заболевания, при котором на коже появляются волдыри и шелушение при малейшем прикосновении — результаты часто сравнивают с ожогами третьей степени.
«Мы никогда не слышали об этой болезни», — сказала г-жа Бэрд.
«Мы с мужем являемся носителями одного и того же рецессивного гена, и мы не знали».
Рецессивные расстройства остаются незамеченными, поскольку не вызывают никаких симптомов у носителя.
Первые шесть месяцев Элиза просто кричала.
Элиза Бэрд в младенчестве в больнице.
«Она испытывала постоянные боли, ей требовалась утренняя и вечерняя смена одежды, включая солевые и отбеливающие ванны, которые занимали примерно три часа три раза в неделю», — сказала г-жа Бэрд.
«Ее пальцы рук и ног переплетались и образовывали стриктуры, кожа в горле становилась настолько плотной из-за рубцовой ткани, что она не могла есть нормальную пищу или в отчаянные времена даже не могла глотать собственную слюну.
«Она будет страдать от ссадин роговицы, что означает, что ее глаза будут закрыты на 3-5 дней в затемненной комнате из-за разрыва роговицы».
У Элизы ухудшалась подвижность, ей требовались повязки и перевязки по всему телу каждый день, и она не могла носить обычную одежду или нижнее белье, потому что даже швы на внутренней стороне одежды покрывали ее кожу волдырями.
Несмотря на все это, сказала мисс Бэрд, ее первенец терпеливо и стойко переносил боль.
«Элиза была потрясающей, она не позволяла своему EB определять себя», — сказала г-жа Бэрд.
«Даже врачи в больнице говорили, что у нее есть умение вовлечь вас и заставить поверить, что она лучше, чем она есть на самом деле. Она довольно хорошо скрывала боль ».
По оценкам, около 1000 человек в Австралии имеют ту или иную форму БЭ и более 500000 человек во всем мире.БЭ не всегда может быть очевидным при рождении. Более легкие случаи заболевания могут проявиться, когда ребенок ползает, ходит, бегает или когда молодые люди становятся более физически активными.
«Детей, живущих с БЭ, часто называют« детьми бабочек », потому что их кожа хрупкая, как крылья бабочки», — сказала г-жа Бэрд.
Болезненные последствия буллезного эпидермолиза.
Она сказала, что тот факт, что БЭ настолько редок, означает, что практикующие врачи могут даже не знать об этом заболевании.
«… Так что врачи общей практики в местном сообществе довольно часто не знают, что это такое, они никогда об этом не слышали».
Но людям с тяжелой формой БЭ часто требуется довольно сильное обезболивающее, которое может быть трудно получить в местной общей врачебной практике.
«Например, моя дочь, помимо прочего, принимала метадон», — сказала г-жа Бэрд.
«Таким образом, вы просто не можете получить эту услугу от терапевта … типы лекарств для снятия боли, которые принимают эти тяжелые пациенты с БЭ, терапевты не чувствуют себя комфортно, выписывая их.
«Таким образом, довольно часто тяжелым пациентам с БЭ действительно необходимо пройти через… крупные больницы. Было бы хорошо, если бы они могли получить доступ к этой поддержке через своего местного терапевта ».
Г-жа Бэрд сказала, что люди с БЭ также часто получают кожные инфекции и требуют длительного приема антибиотиков, что может противоречить текущей медицинской практике ограничения приема антибиотиков.
«Моя дочь довольно много принимала антибиотики, это уменьшило ее болезненность, уменьшились боли, она могла двигаться.Просто ей стало лучше, — сказала мисс Бэрд.
«Мы часто меняли их, чтобы она не сопротивлялась им… Я всегда говорил врачам:« Для меня это здесь и сейчас, а также качество жизни. Об остальном позаботимся позже ».
Восемь месяцев назад, всего за шесть недель до своего 18-летия, Элиза умерла из-за почечной недостаточности, вторичного осложнения, связанного с этим заболеванием.
Симона Бэрд и ее дочь Элиза.
Г-жа Бэрд, которая продолжала свою роль координатора поддержки семьи в Ассоциации исследований дистрофического буллезного эпидермолиза (DEBRA) Австралия , организации по защите и поддержке людей, страдающих БЭ, сказала, что необходимы дополнительные исследования, чтобы найти эффективное лечение состояния.
«Видение DEBRA — создать мир, в котором никто не будет страдать от болезненного генетического заболевания кожи, ЭБ», — сказала она.
«Мы привержены поддержке исследований, направленных на разработку инновационных методов лечения, и однажды мы найдем лекарство.
«Жизнь с ЭБ изменила нашу жизнь, она изменила то, кем мы являемся, но для того, чтобы с нами была Элиза, я бы не стал менять ее для всего мира».
буллезный эпидермолиз генетические нарушения редкие заболевания
НовостиЕженедельный опрос GP Поддерживаете ли вы номера позиций MBS для конкретных заболеваний?
Буллезный эпидермолиз | Детская больница Колорадо
Как лечится буллезный эпидермолиз?
К сожалению, в настоящее время лекарства от буллезного эпидермолиза не существует.Лечение сосредоточено на уходе за ранами и устранении других симптомов БЭ. Важно помнить, что не существует универсального подхода к лечению БЭ. Пациенту могут понадобиться все эти подходы или несколько:
- Частая перевязка для поддержания чистоты и защиты волдырей — очень важный и важный аспект лечения БЭ. Некоторые волдыри также необходимо вскрыть (вскрыть) и осушить.
- Лекарство можно использовать для лечения боли и зуда, связанных с БЭ.
- Принимать ванны с отбеливателем или солью для бассейнов также может утешение пациентов с БЭ.
- В случае проблем с глотанием и образования пузырей / рубцов на пищеводе может быть рассмотрена процедура, называемая дилатацией, с целью расширения пищевода. Если пациент не может набрать вес, можно рассмотреть вопрос о гастростомической трубке, чтобы пациент мог получать адекватное питание.
- Хирургия также может улучшить деформацию рук.
- Приветствуется физиотерапия и трудотерапия для поддержания диапазона движений и внесения изменений в повседневную деятельность.
Почему стоит выбрать Детский Колорадо для лечения БЭ вашего ребенка?
Children’s Colorado предлагает единственный в регионе центр для младенцев, детей и подростков с буллезным эпидермолизом. Наши специально обученные и опытные врачи обеспечивают деликатный индивидуальный уход, который требуется детям. Пациенты обычно принимают участие в физиотерапии и терапии для решения проблем мобильности, потребностей в оборудовании, кормления и функции рук.
Клиника буллезного эпидермолиза
Мы знаем, что нет двух одинаковых случаев БЭ.Мы предлагаем многопрофильную клинику, доступную один раз в месяц, где пациенты посещают клинический персонал из различных отделений Детского Колорадо, включая дерматологию, педиатрию, питание, физиотерапию, трудотерапию, анестезию и социальную работу. Кроме того, доступны консультации стоматологов, специалистов по обезболиванию, психиатрии, гастроэнтерологии и других специалистов, в зависимости от потребностей пациента.
Решения относительно обезболивания во время процедур и обследований принимаются с семьей и командой EB, чтобы определить, что лучше всего для каждого ребенка.Пациенты имеют доступ к службам анестезии и бригаде обезболивающих, чтобы свести к минимуму боль во время процедур и обследований, а также к медсестре, работающей полный рабочий день, которая помогает менять повязки в больнице. Клиника также предоставляет родителям ценную возможность пообщаться и поговорить с другими родителями пострадавших детей.
После каждой клиники группа EB будет координировать уход и оформлять рекомендации по оказанию помощи лечащему врачу вашего ребенка. Полный отчет создается после каждого посещения.
Зимний приключенческий лагерь
Ежегодно клиника EB спонсирует зимний приключенческий лагерь (Camp Spirit) для детей с рецессивно-дистрофическим БЭ.Лагерь проводится в Винтер-парке, штат Колорадо, где участники катаются на би-лыжах, учатся водить собачьи упряжки, кататься на снегоходах и наблюдать за дикой природой. Отдыхающие также могут принять участие в увлекательных вечерних мероприятиях. Для получения дополнительной информации о зимнем лагере приключений, пожалуйста, свяжитесь с дерматологической клиникой по телефону 720-777-8445.
Буллезный эпидермолиз (EB) — Castle Creek Biosciences, Inc.: Castle Creek Biosciences, Inc.
Американская ассоциация исследований дистрофического буллезного эпидермолиза
Американская ассоциация по исследованию буллезного дистрофического эпидермолиза (debra) — единственная национальная некоммерческая организация, занимающаяся финансированием исследований и предоставлением бесплатных услуг и программ для людей с буллезным эпидермолизом (EB).
Castle Creek Biosciences имеет честь поддержать это мероприятие:
Дебра из Америки Видео: История Рафи — не повредит смотреть
Это история Рафи Лили, который живет с буллезным эпидермолизом (БЭ), редким и опасным для жизни заболеванием кожи, которым страдают дети с рождения. Узнайте больше о Rafi and Rafi’s Run здесь.
Партнерство по исследованиям EB
Партнерство EB Research Partnership — крупнейшая некоммерческая организация, занимающаяся финансированием исследований, чтобы дети с EB могли вырасти и жить полноценной, безболезненной жизнью.
Castle Creek Biosciences имеет честь поддерживать эти мероприятия:
- Образовательная программа Европейского совета сообщества на 2021 год
- Погружение для Элоди, виртуальное событие, 28 марта
- Believe in Brady, Houston, Date TBD
- All in for a Cure, Virtual Event, TBD
- Take Flight 5K and Fun Run, Virtual Event, 23 июля — 6 августа
- Изменение для Чарли, Чикаго, октябрь г.
- Venture into Cures, ноябрь г.
Знакомьтесь, Роуэн
Роуэн, 3-летняя девочка, страдающая рецессивным дистрофическим буллезным эпидермолизом.EB Research Partnership дала родителям Роуэна надежду на излечение их дочери и всех остальных, страдающих этим разрушительным заболеванием.
Знакомьтесь, Брэди
Брейди Храбрый и его семья не перестанут сражаться, пока не найдут лекарство.
День редких заболеваний
Rare Disease Day® проводится в последний день февраля каждого года, чтобы привлечь внимание мировой общественности к редким заболеваниям. Узнайте больше об этом особом дне, посвященном тем, кто страдает редкой болезнью, и ее влиянию на их жизнь.
Национальная организация по редким заболеваниям (NORD)
NORD предоставляет единый голос для людей, борющихся с редкими заболеваниями — этих отважных людей, родителей и других опекунов, стремящихся помочь им, так что им не придется сражаться в одиночку.
Глобальные гены
Global Genes ™ — одна из ведущих мировых организаций по защите интересов пациентов с редкими заболеваниями. Их миссия состоит в том, чтобы повышать осведомленность, просвещать мировое сообщество и обеспечивать важные связи и ресурсы, которые позволяют активистам стать активистами своей болезни.
Фонд по борьбе с редкими заболеваниями EveryLife
EveryLife Foundation for Rare Diseases — это некоммерческая организация, деятельность которой направлена на ускорение биотехнологических инноваций для лечения редких заболеваний с помощью государственной политики, основанной на научных исследованиях.
Castle Creek Biosciences имеет честь поддержать это мероприятие:
Альянс педиатрических дерматологических исследований
В ответ на неудовлетворенные потребности в исследованиях в детской дерматологии, требующие совместных многоцентровых усилий, руководители дерматологов создали в 2012 году Альянс педиатрических дерматологических исследований (PeDRA).PeDRA — это исследовательское подразделение Общества детской дерматологии.
Фото: любезно предоставлено Американской ассоциацией исследований дистрофического буллезного эпидермолиза. Используется с разрешения.
Обратите внимание: : Сторонние сайты, перечисленные на этой странице, предназначены только для информационных целей. Castle Creek Biosciences прилагает все усилия, чтобы предложить людям, живущим с буллезным эпидермолизом, самые полезные и точные ресурсы; тем не менее, мы не одобряем и не несем ответственности за содержание этих сторонних веб-сайтов.
Буллезный эпидермолиз — обзор
БУЛЛОЗНЫЙ ЭПИДЕРМОЛИЗ
Буллезный эпидермолиз (БЭ) представляет собой редкую, сложную группу наследственных механобуллезных заболеваний с различной наследственностью, клиническими проявлениями и ультраструктурными и молекулярными дефектами. Есть три основные группы EB и многочисленные сложные схемы подклассификации. Три основные категории — симплексные, соединительные и дистрофические — основаны на уровне эпидермиса или зоны базальной мембраны, на которой образуется волдырь.
EB симплекс вызывается наследственным аутосомно-доминантным дефектом кератинов 5 и 14, который приводит к цитолизу и образованию пузырьков. Хотя при обычной гистологии везикула кажется субэпидермальной, плоскость расщепления фактически находится в цитоплазме базального клеточного слоя. Мельчайшие фрагменты цитоплазмы, все еще прилипшие к дну волдыря, обычно оцениваются с помощью иммуногистохимии против кератина 177 или ультраструктурных методов. Кроме того, на крыше буллы видны дегенеративные изменения базальных клеток, что отсутствует при других вариантах EB.Выявлено несколько типов симплексного EB, включая Weber-Cockayne (локализованный), Koebner (генерализованный), EB simplex herpetiformis (Dowling-Meara), EB simplex herpetiformis с пятнистой пигментацией и EB simplex superficialis.
Junctional EB наследуется как аутосомно-рецессивный признак, связанный с дефектом гемидесмосом и заякоренным комплексом филаментов (ламинин 5 или коллаген типа XVII), что приводит к образованию пузырьков с плоскостью расщепления внутри lamina lucida. Клиническое проявление узловой БЭ варьируется от довольно доброкачественного заболевания до тяжелого состояния, напоминающего дистрофический БЭ (см. Далее).
EB dystrophica может передаваться по наследству как по аутосомно-доминантному, так и по аутосомно-рецессивному признаку. Эта форма EB, которая обычно более тяжелая, чем другие, вызывается дефектом закрепляющих фибрилл (коллаген VII типа), в результате чего образуются субэпидермальные пузырьки с плоскостью расщепления ниже lamina densa. Нарушение базальной мембраны приводит к повторяющимся повреждениям эпидермиса и рубцеванию. Этот постоянный стимул вызывает активированный фенотип эпидермиса с высокой склонностью к развитию плоскоклеточного рака, часто с метастатическим потенциалом и последующей смертью.