
Лежена синдром: Синдром кошачьего крика
Синдром кошачьего крика
Синдром кошачьего крика, или синдром Лежена – это наследственное заболевание, характерной особенностью которого является плач ребёнка, напоминающий крик кошки. Этот синдром относиться к хромосомной патологии, то есть все симптомы вызваны отсутствием части генетического материала, расположенного в коротком плече 5й хромосомы. Выявляется у 1 новорождённого на 45000 – 50000 тысяч детей. Чаще болеют девочки, соотношение примерно 4:3.
Узнать с точностью 99% о риске Синдрома кошачьего крика и других хромосомных аномалий, а также пол плода, можно с 9 недель беременности, всего лишь сдав кровь из вены. Подробнее о НИПТ-тесте.Признаки Синдрома кошачьего крика
- Характерный плач ребёнка, напоминающий мяуканье кошки. Это связано с особенностью строения гортани – она узкая недоразвитая, хрящи тонкие.
- Около 1/3 детей теряют эту характерную черту до 2 лет, у других она остаётся на всю жизнь;
- Низкий вес при рождении (до 2500г) даже при доношенной беременности;
- Нарушено сосание и глотание;
- Обильное слюноотделение;
- Круглое лунообразное лицо.
 С возрастом этот симптом может уменьшиться и лицо приобретает обычные черты;
С возрастом этот симптом может уменьшиться и лицо приобретает обычные черты; - Глаза широкопосажены, раскосые с опущенными наружными углами, у внутреннего угла глаза есть складка – эпикантус;
- Нос широкий с плоской переносицей;
- Уши расположены низко;
- Микроцефалия (малые размеры головного мозга и черепа) с сильно выступающими лобными буграми. С возрастом этот признак выявляется более отчётливо.
- Маленькая нижняя челюсть;
- Короткая шея с кожными складками;
- Особенности поведения, такие как гиперактивность, агрессия, истерики, однообразные движения;
- Снижен тонус мышц всего организма;
- Пороки сердца (дефект межжелудочковой или межпредсердной перегородок, тетрада Фалло)
- Запоры;
 С возрастом этот симптом может уменьшиться и лицо приобретает обычные черты;
С возрастом этот симптом может уменьшиться и лицо приобретает обычные черты;Причины Синдрома кошачьего крика
Спровоцировать мутацию, которая приводит к развитию синдрома кошачьего крика, могут любые повреждающие факторы, воздействующие либо на половые клетки родителей, либо на уже оплодотворённую яйцеклетку во время её дробления и формирования зиготы.
Прогноз заболевания
Продолжительность жизни детей с синдромом кошачьего крика зависит от степени повреждения хромосомы, уровня оказания медицинской помощи и образа жизни. По данным разных источников есть люди, дожившие до 60 лет.
Пренатальная диагностика
1) Инвазивные исследования (амниоцентез, биопсия хориона) в основном назначают тем женщинам, у которых наблюдается повышенный риск того, что родится малыша с Синдромом кошачьего крика, например, пациенткам, чей возраст превышает 35 лет или с плохими результатами неинвазивных тестов: УЗИ и анализов. Инвазивные методы диагностики являются высокоточными, однако, учитывая риск осложнений, не подходят для массового проведения всем беременным, а проводятся только по особым показаниям.
2) Неинвазивные технологии, так называемые скрининги. Скрининг – комплексное исследование беременных женщин на наличие у плода хромосомных аномалий. Выделено несколько признаков, указывающих на высокий риск наличия заболевания, которые может выявить УЗИ плода (отсутствие носовой кости, увеличенная толщина воротникового пространства, недостаточная длина бедренных и плечевых костей и другие особенности).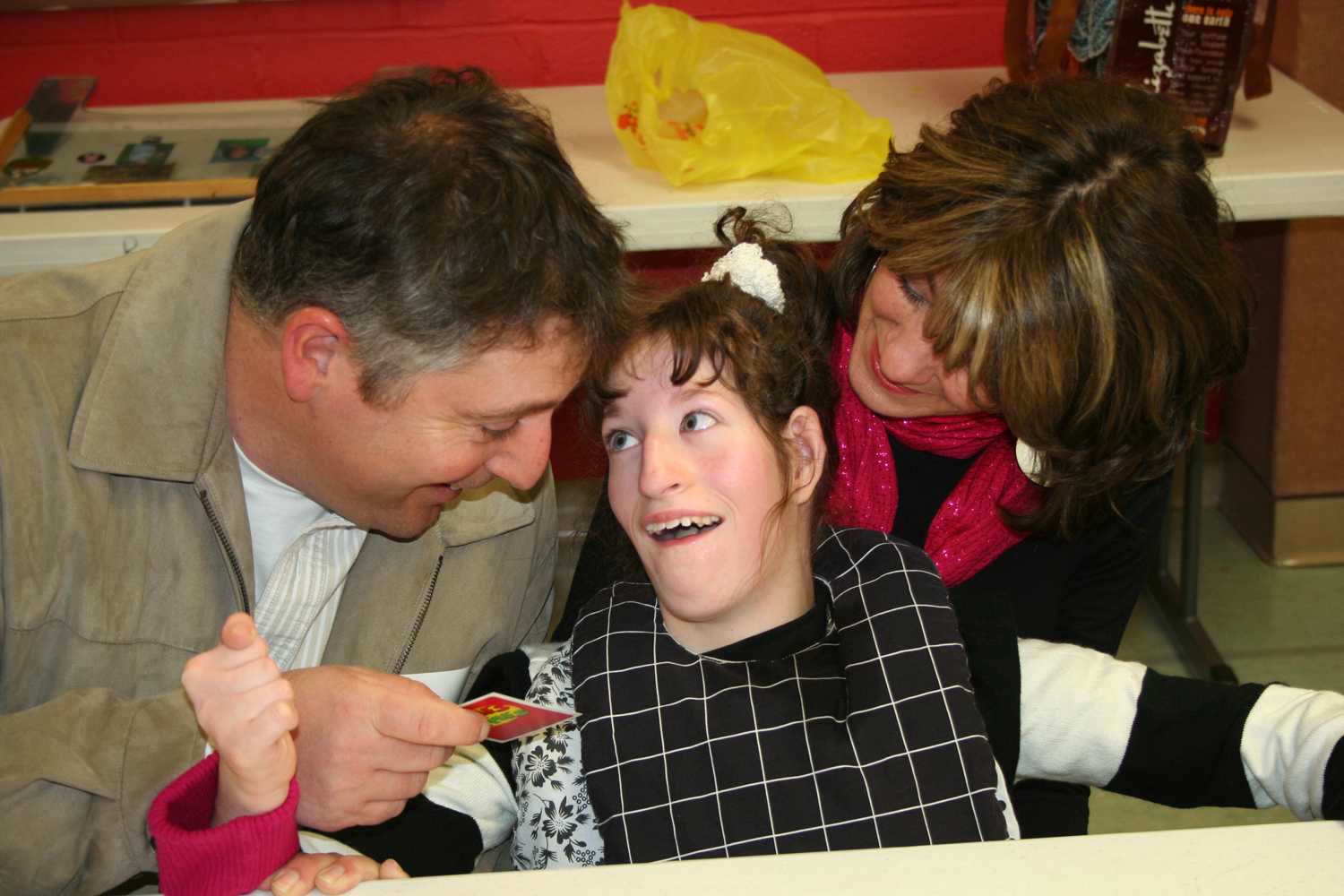 В комплексе с УЗИ идёт биохимический анализ крови матери на такие гормоны как свободный бета-ХГЧ и PAPP-A. Полученные данные по биохимическим маркерам анализируют в совокупности с результатами ультразвукового исследования, а результат всего скрининга представляет собой расчет риска наличия хромосомной аномалии у плода.
В комплексе с УЗИ идёт биохимический анализ крови матери на такие гормоны как свободный бета-ХГЧ и PAPP-A. Полученные данные по биохимическим маркерам анализируют в совокупности с результатами ультразвукового исследования, а результат всего скрининга представляет собой расчет риска наличия хромосомной аномалии у плода.
Однако при использовании стандартных тестов на Синдром кошачьего крика, лишь у 3% женщин, направленных на инвазивную диагностику действительно подтверждается наличие заболевания. В то же время не исключены и ложно-отрицательные результаты, когда скрининг показывает низкий риск, а ребенок рождается с хромосомной патологией.
Неинвазивный метод исследования (НИПТ-тест)- точность 99%, что намного точнее классической диагностики (УЗИ и биохимический скрининг)
- совершенно безопасен, в отличие от инвазивных методик — для забора материала на анализ необходимо просто взять кровь из вены беременной женщины.
- на ранних сроках: анализ можно проводить уже на 9-й неделе беременности.

DIV >
Синдром Лежена — Генетика
Синдром Лежена это наследственное генетическое заболевание, связанное с изменением строения 5-ой хромосомы. Болезнь получила название по имени французского ученого Жерома Лежена в 1963 году. Для этого заболевания характерен необычный детский крик, напоминающий кошачий
Причиной возникновения синдрома Лежена является отсутствие фрагмента 5-ой хромосомы.
Симптомы
— Плач ребенка, напоминающий кошачье мяуканье
— Изменение в строении гортани или ее недоразвитие
— Отставание в развитии ребенка физическом и умственном
— Небольшой вес при рождении
— Пониженный мышечный тонус
— Лунообразное строение лица с широко расположенными глазами
— Врожденные пороки сердца
— Микроцефалия (уменьшенные размеры черепа и головного мозга при норме других частей тела)
— Характерно низкое расположение и деформация ушных раковин
— Выступающие лобные бугры
— Увеличенный клиновидный нос
— Аномалии внутренних органов (сосудов и почек)
— Ярко выраженные сложности с дыханием
— Паховые грыжи
— Малый размер нижней челюсти
— Косоглазие, астигматизм
Отдельные признаки синдрома Лежена имеют возрастные особенности. Исчезает со временем кошачий плач, мышечная гипотония, лунообразное лицо, а вот микроцефалия, косой разрез глаз приобретают более выраженные черты. Усиливается отставание и в психомоторном развитии ребенка. Учащается свистящее шумное дыхание и обостряются заболевания верхних дыхательных путей
Исчезает со временем кошачий плач, мышечная гипотония, лунообразное лицо, а вот микроцефалия, косой разрез глаз приобретают более выраженные черты. Усиливается отставание и в психомоторном развитии ребенка. Учащается свистящее шумное дыхание и обостряются заболевания верхних дыхательных путей
Диагностика
Диагноз ставят исходя из клинических признаков и издающегося «крика котёнка». А лабораторные исследования подтверждают наличие синдрома при потери участка хромосомы 5
Лечение
Лечение синдрома направлено на устранение сопровождающихся симптомов. В первые месяцы жизни следует беречь ребенка от инфекционных заболеваний. Необходимо постоянное наблюдение со стороны педиатра и психоневролога. Врачами рекомендуются средства, стимулирующие психомоторное развитие ребенка, гимнастика и лечебный массаж
Профилактика
Своевременное проведение медико-генетического консультирования в семьях, где уже были дети с синдромом Лежена и проведение исследований на определение кариотипа родителей.
на Урале живет ребенок с редчайшим «Синдромом кошачьего крика»
Фото: Алексей БУЛАТОВ
Двухлетний Андрюша крепко обнимает маму и трется об нее нежной детской щечкой, мурлыча от удовольствия. Оксана Иванова (фамилия изменена) прижимает ребенка к себе, треплет его за волосы и спрашивает:
— Будешь кушать, котенок?
Из этого ласкового обращения сквозит и материнская любовь, и горькое сочувствие: у сына Оксаны в медицинской карточке записан редкий диагноз «Синдром кошачьего крика».
ВРАЧИ УМИЛЯЛИСЬ ТОМУ, КАК АНДРЮША ПЛАЧЕТ
Когда Андрей появился на свет, в родильной палате зазвучало пронзительное… мяуканье! Медики испуганно осмотрели стерильное помещение – откуда здесь кот? И только через несколько секунд поняли – это плачет новорожденный малыш.
— Первые дни после рождения Андрюши врачи и медсестры откровенно хихикали, — вспоминает Оксана. – Подходили и говорили: у вас сын так забавно кричит, как котенок. Но потом перестали умиляться – стало ясно, что Андрей «мяукает» из-за неправильного строения гортани.
«Раньше Андрей почти ничего не ел, — вспоминает мама Оксана Иванова. – Приходилось кормить его молоком чуть ли не из пипетки»
Фото: Алексей БУЛАТОВ
Медики отправили кровь малыша на генетический анализ и, увидев результаты, ахнули. У новорожденного оказался редчайший «синдром кошачьего крика». Мяуканье Андрюши было лишь вершиной айсберга его заболевания – у крошки нашли проблемы с сердцем, почками и развитием.
Дома Ивановы еще пару недель не могли привыкнуть к необычному плачу сына. Но постепенно родители стали различать «мяуканье» по интонациям и сходу понимать, чего хочет их необыкновенный ребенок.
— Раньше Андрей почти ничего не ел, — вспоминает Оксана. – Приходилось кормить его молоком чуть ли не из пипетки, поэтому те редкие моменты, когда он сам просил покушать, были для нас настоящим праздником!
Синдром кошачьего крика или синдром Лежёна (по имени описавшего его в 1963 году французского ученого) — редкое генетическое расстройство, вызываемое отсутствием фрагмента 5-й хромосомы
Фото: Алексей БУЛАТОВ
Лечение Андрея осложнялось тем, что в небольшом поселке Троицкий под Талицей никто и не слышал о «синдроме кошачьего крика». Местные врачи лишь удивленно поднимали брови, увидев в карточке запись о таинственной болезни. Маме зачастую приходилось самой рассказывать медикам о редком генетическом недуге.
Местные врачи лишь удивленно поднимали брови, увидев в карточке запись о таинственной болезни. Маме зачастую приходилось самой рассказывать медикам о редком генетическом недуге.
— Сынишка до сих пор не научился ходить, — вздыхает Оксана. – А ведь ему уже почти два года. Из-за болезни он даже не говорит – синдром замедлил его развитие.
Впрочем, Андрей вполне бойко играет с другими детьми. Правда, яркие пирамидки и гоночные машинки его не особо интересуют. Парнишке больше по душе коробки и пакеты.
— Игрушки у нас лежат в картонных коробочках, — признается Иванова. – Недавно вышла на минутку из комнаты, возвращаюсь – мама дорогая! Все вытряхнул, надел коробку на голову и спрятался… Все время с этими коробками играет.
Друзья у Андрея тоже необычные – мальчонка находит общий язык даже с самыми вредными… животными. Мама до сих пор с содроганием вспоминает, как однажды увидела Андрюшу, увлеченно играющего со своенравным черным котом Барсиком. А ведь Барсик всегда давал сдачи, стоило любому из членов семьи попытаться погладить его.
— Этот кот исцарапал и меня, и старшего сына, — Оксана показывает шрамы на руках. – Поэтому, когда я увидела, что Андрюшка тянется погладить его, здорово испугалась. Но Барсик даже не выпустил когти – признал, что ли…
«Недавно вышла на минутку из комнаты, возвращаюсь – мама дорогая! Все вытряхнул, надел коробку на голову и спрятался… Все время с этими коробками играет», — говорит Оксана Иванова
Фото: Алексей БУЛАТОВ
РЕБЕНОК-КОТЕНОК
Из-за болезни сына мама по-доброму называет его «ребенок-котенок» и даже замечает некоторые сходства с представителями семейства кошачьих. То малыш смешно фыркнет, то замурлычет (это тоже особенность гортани), уютно устроившись у мамы на руках.
— Слава Богу, мяуканье почти прошло, — говорит Оксана. – Еще несколько месяцев и оно исчезнет окончательно. Так происходит у всех носителей этого синдрома. Но это скорее милый дефект, который не представляет опасности. Сейчас основная проблема в том, что мы все еще не ходим и не говорим.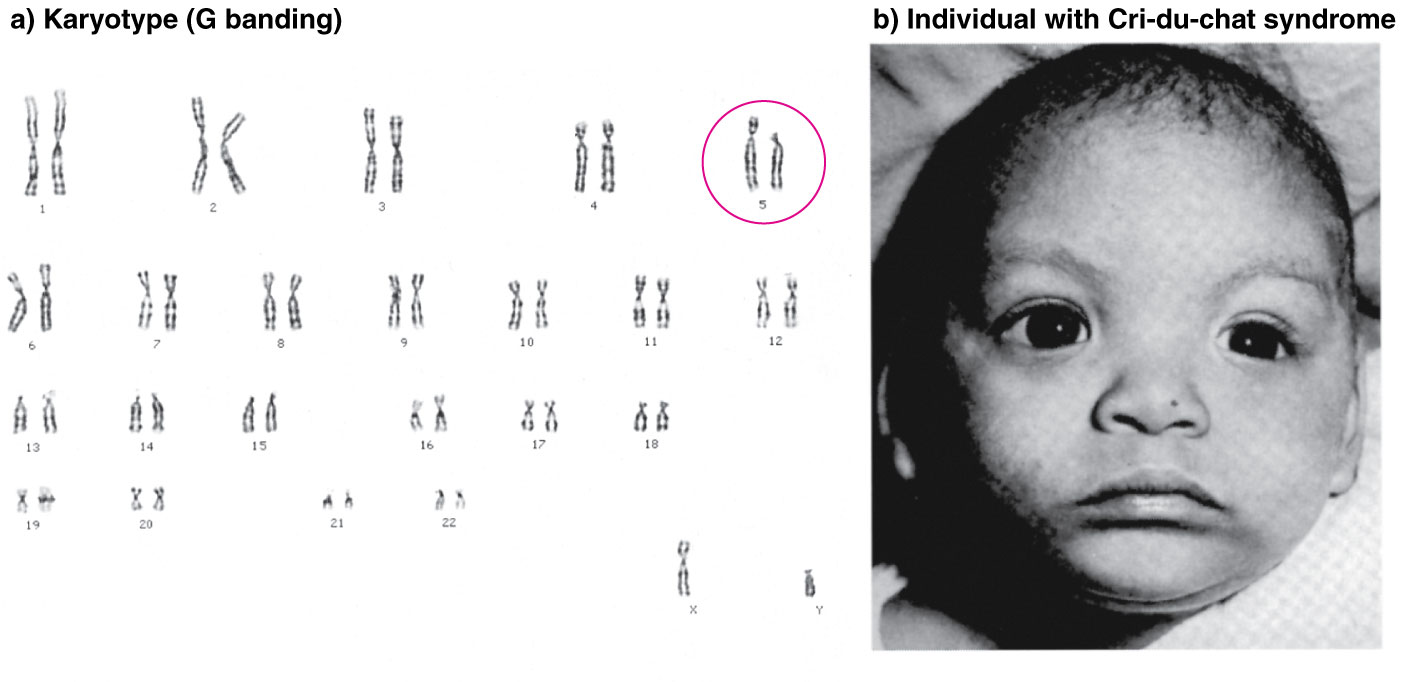 Но мы постоянно занимаемся и не собираемся опускать руки!
Но мы постоянно занимаемся и не собираемся опускать руки!
Чтобы оплачивать лечение сына, отец Андрея стал работать вахтовым методом. Кстати, из-за этого он долго даже не подозревал, что ребенку поставили редкий диагноз.
— Муж узнал о болезни Андрюши, когда ему исполнилось 9 месяцев, — признается Оксана. – До этого я боялась рассказывать ему: сами знаете, многие мужчины бросают семьи. Но наш папа оказался совсем другим.
Евгений, узнав о диагнозе, мигом вернулся с вахты. Крепко обнял любимую, взял на руки Андрея и твердо сказал:
— Будем бороться!
Сейчас отец снова далеко от семьи – работает, чтобы обеспечить лечение своему сынишке. Но лишних денег в семье с ребенком-инвалидом не бывает никогда. Сейчас, например, Андрюше нужны ходунки, фиксирующие тело – без них малышу очень сложно учиться ходить.
Если у вас есть ненужные ходунки, вы можете позвонить в редакцию по номеру +7 (343) 379-27-72 или перевести удобную для вас сумму на номер карты 639002169035760574 (Сбербанк).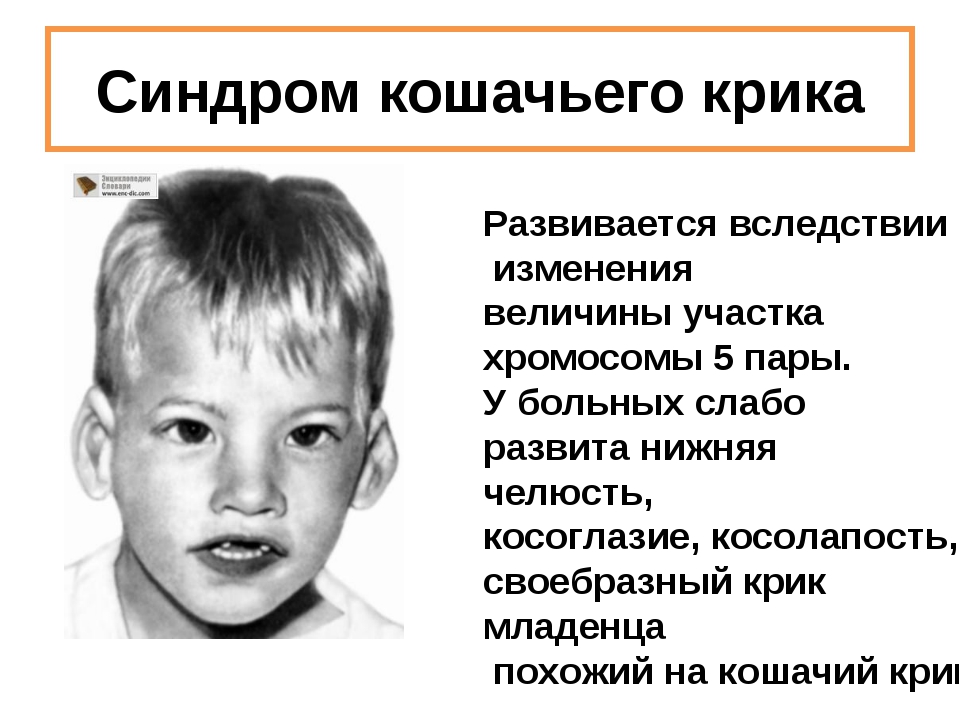
Комментарий эксперта
«Вылечить этот синдром невозможно»
— Синдром кошачьего крика — генетическое заболевание, — объясняет специалист лаборатории «Геномед» Ирина Акимова. — Помочь ребенку с подобной патологией можно только, тщательно ухаживая за ним, ведь подобное изменение (в случае синдрома кошачьего крика – отсутствие хромосомы 5) произошло в каждой клетке организма, и вылечить это, к сожалению, невозможно.
Профилактика синдрома Лежена — МегаЛекции
Синдром кошачьего крика
Синдром «кошачьего крика»–хромосомное нарушение, обусловленное отсутствием фрагмента короткого плеча 5-ой хромосомы. Синдром «кошачьего крика» относится к числу редких хромосомных заболеваний с популяционной частотой 1:45-50 тыс. Среди новорожденных с синдромом «кошачьего крика» отмечается преобладание девочек над мальчиками в соотношении 4:3. Заболевание было описано в 1963 г. французским генетиком и педиатром Ж.
Причины синдрома «кошачьего крика»
Много факторов могут воздействовать на половой клеточный уровень, яйцеклетку, чаще всего заболевание спровоцировано:
1. Наследственными причинами, когда в семье кто-то имел такое заболевание.
2. Вредными привычками. Когда беременная женщина злоупотребляла алкогольными напитками и много курила.
3. Приемом наркотиков, из-за них внутренние органы постепенно разрушаются, затем поражается генетический клеточный аппарат.
4. Воздействием радиации.
3. Употреблением сильнодействующих медикаментозных препаратов, которые женщина принимает в начале беременности
Симптомы синдрома «кошачьего крика»
Дети с данным заболеванием рождаются своевременно, женщина до конца вынашивает ребенка, но он имеет небольшую массу тела, в среднем около 2500 грамм.
Ребенок плачет так, как мяукает кошка, это сразу заметно. Такой звук объясняется тем, что малыш имеет своеобразную анатомическую гортанную особенность. В гортани просвет является узким, а хрящи размягченные. Дети при рождении имеют небольшой надгортанник. Часто синдром проходит уже в 2 года, опасно, когда заболевание остается на всю жизнь.
У многих пациентов наблюдаются врожденные пороки сердца, пороки развития почек . Реже отмечается запоры, кишечная непроходимость.
Синдром кошачьего крика и аутизм
Когда врач начинает исследовать геному семьи, он узнает, что в роду у ребенка был больной аутизмом. Если у кого-то из членов семьи были аномальные явления в хромосомном наборе, заболевание будет прогрессировать.
Дети, у которых синдром Лежена отличаются лунообразным лицом. При синдроме деформируются ушные раковины, укорачивается шея. Больной ребенок имеет заболевание – микроцефалия, у него проблемы с рефлексами, наблюдается гипотония в мышцах, нарушается сосание груди, глотание молока.
При синдроме деформируются ушные раковины, укорачивается шея. Больной ребенок имеет заболевание – микроцефалия, у него проблемы с рефлексами, наблюдается гипотония в мышцах, нарушается сосание груди, глотание молока.
Иногда больной синдромом кошачьей крик имеет серьезные глазные заболевания — косоглазие, проблемы со зрительным нервом, катаракту. У ребенка наблюдаются патологические процессы в опорно-двигательном аппарате – вывихнуто бедро, грыжа паха и пупка, синдактилия стоп. Опасно, когда синдром сопровождается заболеваниями почек, сердца, сосудов, желудка, кишечника. Дети, у которых синдром кошачий крик гиперактивны, страдают от истерии, агрессивны. Часто у них возникают проблемы с физическим развитием, ребенок умственно отсталый, имеет проблемы с речью.
Часто больные дети живут недолго, из-за того, что синдром Лежена сопровождается серьезными патологическими процессами в жизненно важных органах. Реже ребенок может дожить до 15 лет, и только 5% может жить с заболеванием около 50 лет
Лечение синдрома Лежена
Современная медицина до сих пор не имеет специальных методов лечения хромосомного заболевания. Курс терапии может быть симптоматическим, он помогает поддержать в норме состояние системных органов и продолжить жизнь больного.
Курс терапии может быть симптоматическим, он помогает поддержать в норме состояние системных органов и продолжить жизнь больного.
Чтобы наладилась психомоторная функция у ребенка, нужно обратиться к неврологу, пройти дополнительно медикаментозную терапию, дополнительно нужна консультация психолога. Дополнительно ребенка нужно показать логопеду, физиотерапевту, дефектологу.
Если ребенок родился с кошачьим синдромом, имеет врожденные пороки сердца, необходима срочная операция. Часто синдром сопровождается аномалиями в мочевыделительной системе, поэтому сразу нужно обращаться к урологу.
Из-за того, что у ребенка общий гипотонус, ему нужен постоянный массаж, лечебная гимнастика. Врачи выписывают лекарственные препараты, с помощью которых происходит стимуляция психомоторного развития
Прогноз при синдроме кошачьего крика
Сколько будет жить ребенок, зависит от того, насколько повреждены хромосомы. Очень редко у человека все нормально и он доживает до 60 лет.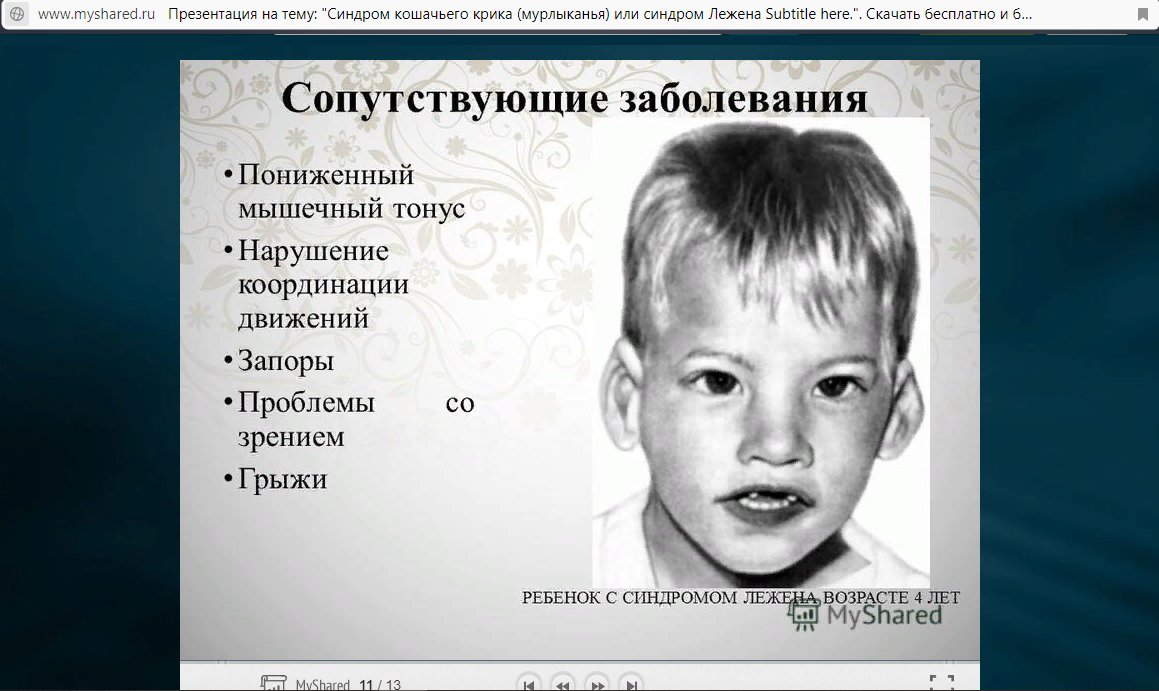 Если заниматься ребенком, можно его научить чтению, писанию, адаптироваться к окружающей среде.
Если заниматься ребенком, можно его научить чтению, писанию, адаптироваться к окружающей среде.
Продолжительность жизни с данным заболеванием зависит от того, какие врожденные пороки имеет ребенок, оказана ли своевременно медицинская, психологическая помощь. Часто, если заниматься с ребенком, он будет иметь в словарном запасе несколько предложений.
Профилактика синдрома Лежена
Необходимо тщательно готовиться к беременности, сдать все необходимые анализы, чтобы исключить все генетические причины, также беречься во время беременности, отказаться от всех вредных привычек, защищать себя и ребенка от всех неблагоприятных факторов. Если в семье рождается ребенок с синдромом кошачий крик, необходимо дополнительно сдать цитогенетический анализ, чтобы исключить все осложнения.
Итак, синдром кошачьего крика – это редкое, но опасное заболевание. Лучше предотвратить его, потому что вылечить невозможно. Будущая мама должна тщательно относиться к своему здоровью, забыть за алкоголь, курение.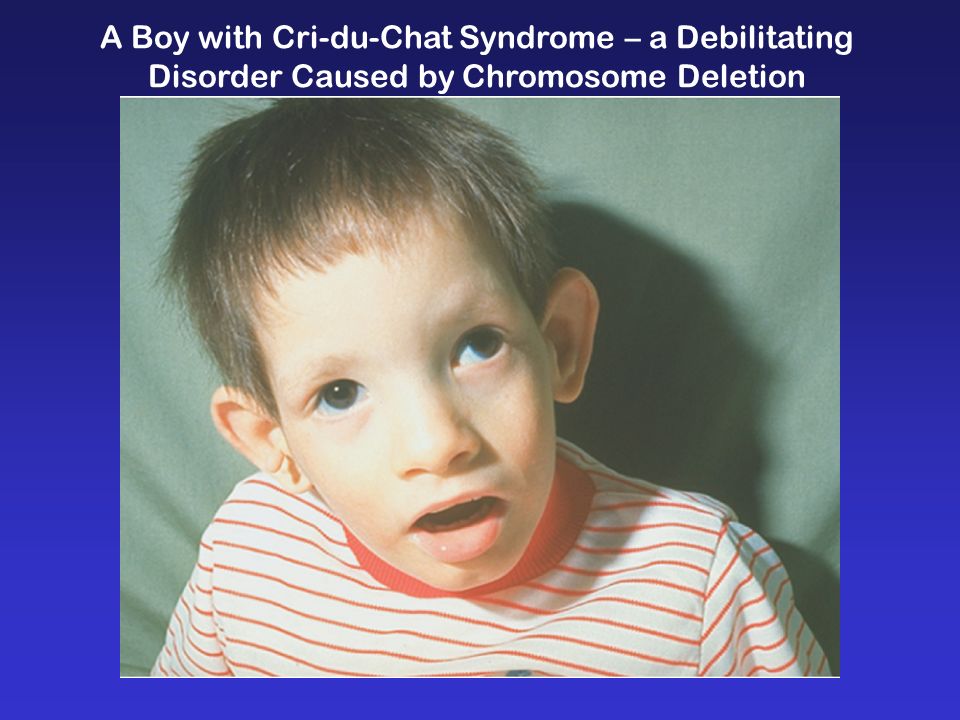 Если ребенок родился с аномалиями, тщательно следить за его здоровьем, обратить, полностью обследовать его, потому что часто синдром сопровождается разными патологиями в органах.
Если ребенок родился с аномалиями, тщательно следить за его здоровьем, обратить, полностью обследовать его, потому что часто синдром сопровождается разными патологиями в органах.
Рекомендуемые страницы:
Воспользуйтесь поиском по сайту:
Синдром Жубера, Анализ числа копий гена NPHP1
Метод определения ПЦР, секвенирование
Исследуемый материал Цельная кровь (с ЭДТА)
Анализ числа копий гена NPHP1.
Гены, ответственные за развитие заболевания.
Ген NPHP1 (NEPHROCYSTIN 1) расположен на хромосоме 2 в регионе 2q13. Содержит 20 экзонов.
Мутации в данном гене приводят также к развитию ювенильного нефронофтиза, тип 1. К развитию синдрома Жубера приводят также мутации в гене AHI1.
Определение заболевания.
Редкое генетическое заболевание, сопровождающееся недоразвитием или отсутствием червя мозжечка, управляющего балансом и координацией.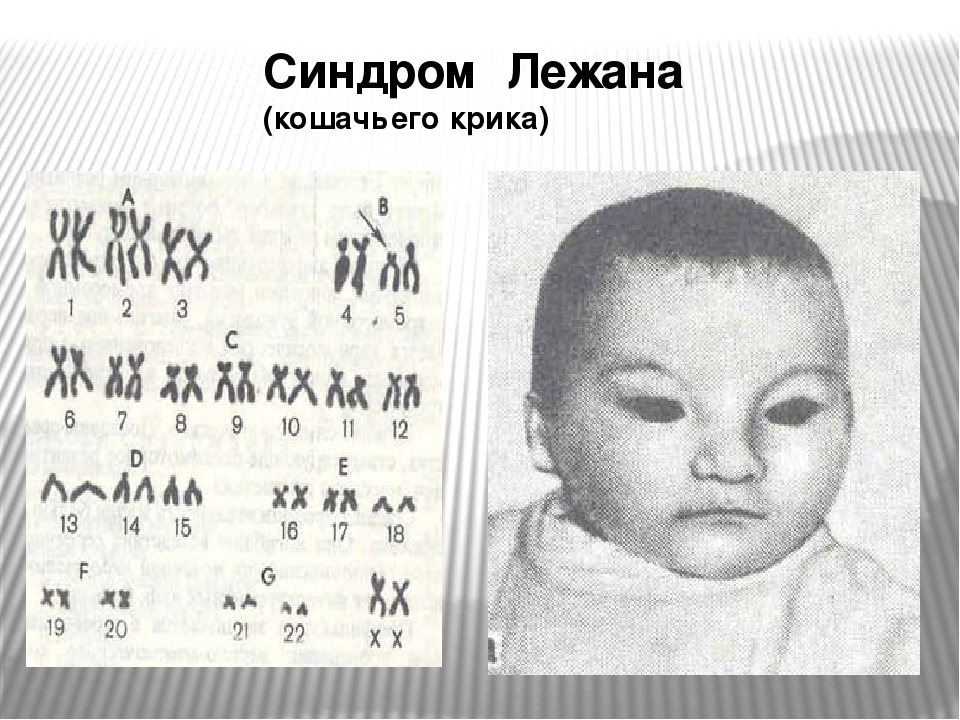
Патогенез и клиническая картина.
Из-за плохо сформированного ствола мозга могут наблюдаться нарушения дыхания вплоть до остановки дыхания, ненормально учащенное дыхание. Наблюдаются неправильные хаотичные движения глаз и языка. У новорожденных может отмечаться пониженный мышечный тонус. Проявляется также задержкой психомоторного развития. Бывает нефронофтиз.
Частота встречаемости: не установлена. Заболевание редкое.
Перечень исследуемых мутаций может быть предоставлен по запросу.
Литература
- Joubert, M., Eisenring, J. J., Robb, J. P., Andermann, F. Familial agenesis of the cerebellar vermis: a syndrome of episodic hyperpnea, abnormal eye movements, ataxia, and retardation. Neurology 19: 813-825, 1969. Note: Reprinted in J. Child Neurol. 14: 554-564, 1999.
- Parisi, M. A., Bennett, C. L., Eckert, M. L., Dobyns, W. B., Gleeson, J. G., Shaw, D. W. W., McDonald, R., Eddy, A., Chance, P. F., Glass, I. A. The NPHP1 gene deletion associated with juvenile nephronophthisis is present in a subset of individuals with Joubert syndrome. Am. J. Hum. Genet. 75: 82-91, 2004.
- OMIM.
 A. The NPHP1 gene deletion associated with juvenile nephronophthisis is present in a subset of individuals with Joubert syndrome. Am. J. Hum. Genet. 75: 82-91, 2004.
A. The NPHP1 gene deletion associated with juvenile nephronophthisis is present in a subset of individuals with Joubert syndrome. Am. J. Hum. Genet. 75: 82-91, 2004. ЛЕЖЕНА СИНДРОМ — это 📕 что такое ЛЕЖЕНА СИНДРОМ
врожденный комплекс пороков развития, обусловленный нарушением структуры одной из хромосом группы В. Описан в 1963 г. Леженом с соавторами.
Частота синдрома среди новорожденных около 1: 3000, мальчики и девочки поражаются одинаково часто. Зависимость частоты рождения детей с Л.с. от возраста родителей не установлена. Синдром обусловлен изменениями короткого плеча хромосомы 5-й пары (рис. 1), возникающими чаще вследствие потери участка хромосомы, реже — структурной перестройки хромосомы или перемещения сегмента хромосомы внутри хромосомного набора. Описаны и другие варианты сбалансированных транслокаций в клетках родителей, приводившие к рождению детей с синдромом Лежена. Некоторая вариабельность клинических проявлений синдрома, по-видимому, зависит от размеров недостающего участка хромосомы.
Дети с Л.с. обычно рождаются с низкой массой тела (до 2500 г) даже при доношенной беременности. Наиболее постоянным признаком является специфический плач, напоминающий кошачье мяуканье. Этот симптом обусловлен особенностью строения гортани, определяемым при ларингоскопии — маленьким вялым надгортанником, который может опускаться над голосовой щелью. Голосовые складки не изменены. Рентгенологически отмечается уменьшение воздушного пространства над голосовыми складками.
В раннем детском возрасте характерны лунообразное лицо, косой разрез глаз с опущенными наружными углами, эпикантус (складка у внутреннего угла глаза), гипертелоризм (широко расставленные глаза), несколько уплощенный нос, низко расположенные ушные раковины (рис. 2), впереди которых часто имеются небольшие (размером 1—3 мм) круглые фиброзные узелки. Мозговой череп относительно малых размеров (микроцефалия), долихоцефальной формы (значительное преобладание продольных размеров над поперечными) или с выступающими лобными буграми. Отмечается маленькая нижняя челюсть и короткая шея с избыточной кожей, формирующей крыловидные складки. В некоторых случаях отмечается расщепление верхней губы или неба либо высокое готическое небо и расщепление язычка. Возможны преходящее или постоянное Косоглазие, астигматизм (см. Рефракция глаза). В ряде случаев выявляют изменения глазного дна, в частности очаги депигментации сетчатки, а также атрофию зрительного нерва. Из аномалий развития внутренних органов наиболее часты пороки развития сердца и сосудов, почек. У мальчиков часто бывает Гипоспадия. Может быть четырехпалость или короткая, треугольной формы средняя фаланга V пальца. Общая мышечная гипотония, характерная для новорожденных с Л.с., обычно сохраняется в течение 1 года и дольше. У всех детей с Л.с. наблюдается умственная отсталость, большинство отстают в физическом развитии. Биохимические нарушения при Л.с. неспецифичны: длительное сохранение фетального гемоглобина, некоторое снижение содержания альбумина в сыворотке крови, умеренная аминоацидемия и аминоацидурия.
Отмечается маленькая нижняя челюсть и короткая шея с избыточной кожей, формирующей крыловидные складки. В некоторых случаях отмечается расщепление верхней губы или неба либо высокое готическое небо и расщепление язычка. Возможны преходящее или постоянное Косоглазие, астигматизм (см. Рефракция глаза). В ряде случаев выявляют изменения глазного дна, в частности очаги депигментации сетчатки, а также атрофию зрительного нерва. Из аномалий развития внутренних органов наиболее часты пороки развития сердца и сосудов, почек. У мальчиков часто бывает Гипоспадия. Может быть четырехпалость или короткая, треугольной формы средняя фаланга V пальца. Общая мышечная гипотония, характерная для новорожденных с Л.с., обычно сохраняется в течение 1 года и дольше. У всех детей с Л.с. наблюдается умственная отсталость, большинство отстают в физическом развитии. Биохимические нарушения при Л.с. неспецифичны: длительное сохранение фетального гемоглобина, некоторое снижение содержания альбумина в сыворотке крови, умеренная аминоацидемия и аминоацидурия.
Частота и выраженность отдельных признаков Л.с. имеют возрастную зависимость. Такие признаки, как плач, напоминающий кошачье мяуканье, мышечная гипотония, лунообразное лицо в большинстве случаев с возрастом полностью исчезают, а микроцефалия, косой разрез глаз становятся более выраженными; прогрессирует отставание в психомоторном развитии. Может быть стридор; больные подвержены заболеваниям верхних дыхательных путей.
Синдром дифференцируют с другими врожденными пороками развития хромосомной и нехромосомной этиологии. Диагноз подтверждается кариологическим исследованием с применением одного из методов идентификации хромосом.
Лечение симптоматическое. Показаны средства, стимулирующие психомоторное развитие, лечебный массаж и гимнастика.
Средняя продолжительность жизни больных снижена. Они погибают вследствие сердечной недостаточности (Сердечная недостаточность) или почечной недостаточности (Почечная недостаточность), от интеркуррентных инфекционных болезней.
Профилактика заключается в своевременном проведении медико-генетического консультирования (Медико-генетическое консультирование) в семьях, где имелись больные с синдромом Лежена и основывается на определении кариотипа родителей, у которых был больной ребенок. Наличие изменений короткого плеча 5-й пары хромосом является абсолютным показанием для антенатального определения кариотипа плода при последующих беременностях путем амниоцентеза и исследования амниотических клеток. Сбалансированная транслокация у одного из родителей требует также исследования кариотипа у его кровных родственников с целью выявления лиц, имеющих транслокацию.
Библиогр.: Козлова С.И. и др. Наследственные синдромы и медико-генетическое консультирование, с. 337, М., 1987; Маринчева Г.С. и Гаврилов В.И. Умственная отсталость при наследственных болезнях, с. 180, М., 1988; Тератология человека, под ред. Г.И. Лазюка, с. 314, М., 1979.
набор больной с синдромом Лежена: групповая (от А до G) и индивидуальная идентификация хромосом (стрелкой указан дефект короткого плеча хромосомы 5-й пары, вторая хромосома не изменена)»>Рис. 1. Хромосомный набор больной с синдромом Лежена: групповая (от А до G) и индивидуальная идентификация хромосом (стрелкой указан дефект короткого плеча хромосомы 5-й пары, вторая хромосома не изменена).
1. Хромосомный набор больной с синдромом Лежена: групповая (от А до G) и индивидуальная идентификация хромосом (стрелкой указан дефект короткого плеча хромосомы 5-й пары, вторая хромосома не изменена).
Рис. 2а). Ребенок с синдромом Лежена в возрасте 4 дней: лунообразное лицо, косой разрез глаз с опущенными наружными углами, несколько уплощенный нос, низко расположенные ушные раковины.
Рис. 2б). Ребенок с синдромом Лежена возрасте 4 лет.
1
Первый слайд презентации: Синдром кошачьего крика (Синдром Лежена )
Изображение слайда
2
Слайд 2
Синдром кошачьего крика, или синдром Лежена – это наследственное заболевание, характерной особенностью которого является плач ребёнка, напоминающий крик кошки. Этот синдром относиться к хромосомной патологии
Этот синдром относиться к хромосомной патологии
Изображение слайда
3
Слайд 3
Впервые синдром кошачьего крика был описан в шестидесятых годах XX века французским генетиком-Леженом. Именно он дал название данной патологии, поскольку плач больного ребёнка очень похож на крик кота.
Изображение слайда
4
Слайд 4
Тип мутации:
Генетически данный синдром объясняют частичной моносомией. Его возникновение связано с хромосомной перестройкой, в процессе которой теряется от 1/3 до половины из p-плеча хромосомы №5, которая содержит в себе около 6% всего генетического материала. На клиническую картину заболевания влияет не то, какова величина исчезнувшей части, а то, какой именно фрагмент был утрачен.
Его возникновение связано с хромосомной перестройкой, в процессе которой теряется от 1/3 до половины из p-плеча хромосомы №5, которая содержит в себе около 6% всего генетического материала. На клиническую картину заболевания влияет не то, какова величина исчезнувшей части, а то, какой именно фрагмент был утрачен.
Изображение слайда
5
Слайд 5
Чем обусловлен Обусловлен изменением гортани: сужением мягкости хрящей, отечностью или необычной складчатостью слизистой оболочки, уменьшением надгортанника
Изображение слайда
6
Слайд 6
Основные причины:
Наследственный фактор. Если в семье уже есть ребёнок с данным синдромом, то вероятность рождения второго с таким же заболеванием очень высока;
Алкоголь и курение оказывают повреждающее действие на все клетки организма, но наиболее чувствительны к нему половые клетки и зародыш;
Наркотические вещества разрушают организм изнутри, поражая генетический аппарат клеток;
Действие ионизирующей радиации;
Сильнодействующие химические вещества и лекарственные препараты;
Если в семье уже есть ребёнок с данным синдромом, то вероятность рождения второго с таким же заболеванием очень высока;
Алкоголь и курение оказывают повреждающее действие на все клетки организма, но наиболее чувствительны к нему половые клетки и зародыш;
Наркотические вещества разрушают организм изнутри, поражая генетический аппарат клеток;
Действие ионизирующей радиации;
Сильнодействующие химические вещества и лекарственные препараты;
Изображение слайда
7
Слайд 7: В основе синдрома лежат изменения короткого плеча хромосомы 5-й пары, возникающие вследствие делеции (это потеря части хромосомы в результате ее разрыва). При расхождении хромосом в разные клетки в первом делении мейоза хромосома, утратившая часть генетического материала, оказывается несбалансированной
Изображение слайда
8
Слайд 8: Основные признаки синдрома кошачьего крика:
Характерный плач ребёнка, напоминающий мяуканье кошки. Низкий вес при рождении (до 2500г)
Нарушено сосание и глотание;
Обильное слюноотделение ;
Круглое лунообразное лицо.
Глаза широкопосажены
Нос широкий с плоской переносицей ;
Малые размеры головного мозга и черепа с сильно выступающими лобными буграми.
Маленькая нижняя челюсть;
Короткая шея с кожными складками ;
Умственная отсталость, задержка развития речевых и физических навыков
Низкий вес при рождении (до 2500г)
Нарушено сосание и глотание;
Обильное слюноотделение ;
Круглое лунообразное лицо.
Глаза широкопосажены
Нос широкий с плоской переносицей ;
Малые размеры головного мозга и черепа с сильно выступающими лобными буграми.
Маленькая нижняя челюсть;
Короткая шея с кожными складками ;
Умственная отсталость, задержка развития речевых и физических навыков
Изображение слайда
9
Слайд 9: Частота встречаемости
Частота встречаемости составляет 1/40-50тыс новорожденных. Девочки подвержены больше, чем мальчики.
Изображение слайда
10
Последний слайд презентации: Синдром кошачьего крика (Синдром Лежена )
Изображение слайда
Синдром Ли: MedlinePlus Genetics
Синдром Ли может быть вызван мутациями в одном из более чем 75 различных генов. У людей большинство генов находится в ДНК в ядре клетки, называемой ядерной ДНК. Однако некоторые гены находятся в ДНК в специализированных структурах клетки, называемых митохондриями. Этот тип ДНК известен как митохондриальная ДНК (мтДНК). В то время как у большинства людей с синдромом Ли есть мутация в ядерной ДНК, около 20 процентов имеют мутацию в мтДНК.
У людей большинство генов находится в ДНК в ядре клетки, называемой ядерной ДНК. Однако некоторые гены находятся в ДНК в специализированных структурах клетки, называемых митохондриями. Этот тип ДНК известен как митохондриальная ДНК (мтДНК). В то время как у большинства людей с синдромом Ли есть мутация в ядерной ДНК, около 20 процентов имеют мутацию в мтДНК.
Большинство генов, связанных с синдромом Ли, вовлечены в процесс производства энергии в митохондриях.Митохондрии используют кислород для преобразования энергии пищи в форму, которую клетки могут использовать в процессе, называемом окислительным фосфорилированием. В этом процессе участвуют пять белковых комплексов, каждый из которых состоит из нескольких белков. Комплексы называются комплексом I, комплексом II, комплексом III, комплексом IV и комплексом V. Во время окислительного фосфорилирования белковые комплексы управляют выработкой аденозинтрифосфата (АТФ), основного источника энергии клетки, посредством пошагового процесса. перенос отрицательно заряженных частиц, называемых электронами.Многие мутации генов, связанные с синдромом Ли, влияют на белки в этих комплексах или нарушают их сборку. Эти мутации снижают или устраняют активность одного или нескольких из этих комплексов, что может привести к синдрому Ли.
перенос отрицательно заряженных частиц, называемых электронами.Многие мутации генов, связанные с синдромом Ли, влияют на белки в этих комплексах или нарушают их сборку. Эти мутации снижают или устраняют активность одного или нескольких из этих комплексов, что может привести к синдрому Ли.
Нарушение комплекса I, также называемого НАДН: убихинон оксидоредуктаза, является наиболее частой причиной синдрома Ли, на которую приходится почти треть случаев этого состояния. По крайней мере 25 генов, участвующих в образовании комплекса I, обнаруженных в ядерной или митохондриальной ДНК, были связаны с синдромом Ли.
Нарушение комплекса IV, также называемого цитохром с оксидазой или ЦОГ, также является частой причиной синдрома Ли, лежащего в основе примерно 15 процентов случаев. Один из наиболее часто мутирующих генов при синдроме Ли — SURF1 . Этот ген, который находится в ядерной ДНК, дает инструкции по созданию белка, который помогает собрать белковый комплекс ЦОГ (комплекс IV). Этот комплекс, который участвует в последнем этапе переноса электрона при окислительном фосфорилировании, обеспечивает энергию, которая будет использоваться на следующем этапе процесса для генерации АТФ.Мутации в гене SURF1 обычно приводят к аномально короткому белку SURF1, который расщепляется в клетках, что приводит к отсутствию функционального белка SURF1. Потеря этого белка снижает образование нормальных комплексов ЦОГ, что ухудшает выработку энергии митохондриями.
Этот комплекс, который участвует в последнем этапе переноса электрона при окислительном фосфорилировании, обеспечивает энергию, которая будет использоваться на следующем этапе процесса для генерации АТФ.Мутации в гене SURF1 обычно приводят к аномально короткому белку SURF1, который расщепляется в клетках, что приводит к отсутствию функционального белка SURF1. Потеря этого белка снижает образование нормальных комплексов ЦОГ, что ухудшает выработку энергии митохондриями.
Наиболее частая мутация мтДНК при синдроме Ли затрагивает ген MT-ATP6 , который предоставляет инструкции по созданию части комплекса V, также известного как белковый комплекс АТФ-синтазы.Используя энергию других белковых комплексов, АТФ-синтазный комплекс генерирует АТФ. MT-ATP6 Мутации гена, обнаруженные примерно у 10 процентов людей с синдромом Ли, блокируют выработку АТФ. Другие мутации мтДНК, связанные с синдромом Ли, снижают активность других белковых комплексов окислительного фосфорилирования или приводят к уменьшению образования митохондриальных белков, которые нарушают выработку митохондриальной энергии.
Другие мутации гена, связанные с синдромом Ли, снижают активность одного или нескольких белковых комплексов окислительного фосфорилирования или влияют на дополнительные стадии, связанные с выработкой энергии.Например, синдром Ли может быть вызван мутациями в генах, которые образуют комплекс пируватдегидрогеназы или кофермент Q10, оба из которых участвуют в выработке энергии митохондриями. Мутации в генах, которые управляют репликацией мтДНК или выработкой митохондриальных белков, также могут нарушать выработку митохондриальной энергии.
Хотя точный механизм неясен, исследователи полагают, что нарушение окислительного фосфорилирования может привести к гибели клетки из-за уменьшения энергии, доступной в клетке.Некоторые ткани, которым требуется большое количество энергии, такие как мозг, мышцы и сердце, кажутся особенно чувствительными к снижению клеточной энергии. Смерть клеток в головном мозге, вероятно, вызывает характерные поражения, наблюдаемые при синдроме Ли, которые вносят свой вклад в признаки и симптомы этого состояния. Гибель клеток в других чувствительных тканях также может способствовать развитию синдрома Ли.
Гибель клеток в других чувствительных тканях также может способствовать развитию синдрома Ли.
Синдром Ли — NORD (Национальная организация по редким заболеваниям)
УЧЕБНИКИ
Чакраборти П., Фейгенбаум А., Робинсон Б.Дефицит цитохромоксидазы человека. Руководство NORD по редким заболеваниям. Филадельфия, Пенсильвания: Липпинкотт Уильямс и Уилкинс: 2003: 436.
Lyon G, Adams RD, Kolodny EH. Ред. Неврология наследственных болезней обмена веществ в детстве. 2-е изд. Нью-Йорк, штат Нью-Йорк: компании McGraw-Hill; 1996: 94-9.
СТАТЬИ ИЗ ЖУРНАЛА
Несбитт В., Моррисон П.Дж., Крушелл Э. и др. Клинический спектр мутации m.10191T> C при комплексном I-дефицитном синдроме Ли. Dev Med Child Neurol.2012, В печати. PMID: 22364517.
Tuppen HA, Hogan VE, He L, et al. Мутация p.M292T NDUFS2 вызывает комплексный I-дефицитный синдром Ли во многих семьях. Мозг. 2010; 133 (10): 2952-63.
Фридман С.Д., Шоу Д. В., Исхак Г., Гропман А.Л., Сането Р.П. Использование нейровизуализации в диагностике митохондриальных заболеваний. Dev Disabil Res Rev.2010; 16 (2): 129-35.
В., Исхак Г., Гропман А.Л., Сането Р.П. Использование нейровизуализации в диагностике митохондриальных заболеваний. Dev Disabil Res Rev.2010; 16 (2): 129-35.
van Riesen AK и др., Сегментарная дисомия матери при синдроме Ли с дефицитом цитохром с оксидазы, вызванным гомозиготной мутацией SURF1.Нейропедиатрия. 2006; 37: 88-94.
Шифф М., Мине М., Бривет М. и др. Болезнь Ли, вызванная новой мутацией в гене PDHX. Энн Нейрол. 2006; 59 (4): 709-14.
Ван Малдергем Л., Трайбельс Ф., ДиМауро С. и др. Коэнзим Q-зависимая энцефалопатия Ли у двух сестер. Энн Нейрол. 2002; 52 (6): 750-4.
Макино М., Хораи С., Гото Ю., Нонака И. Мутации митохондриальной ДНК при синдроме Ли и их филогенетические последствия. J Hum Genet. 2000; 45 (2): 69-75.
Торберн DR.Синдом Ли: клинические особенности, биохимические и ДНК-аномалии. Митохондриальные новости. 1998; 3: 1, 7-10.
Рахман С. и др., Синдром Ли: клинические особенности, биохимические и ДНК-аномалии. Энн Нейрол. 1996; 39: 343-51.
1996; 39: 343-51.
Santorelli FM, Мутация на участке 8993 митохондриальной ДНК является частой причиной синдрома Ли. Энн Нейрол. 1993; 34: 827-34.
Мэтьюз П.М. и др., Молекулярно-генетическая характеристика Х-сцепленной формы синдрома Ли. Энн Нейрол.1993; 33: 652-5.
Ciafaloni E, et al., Унаследованный от матери синдром Ли. J Pediatr. 1993; 122: 419-22.
Macaya A и др., Нарушения движения при синдроме Ли. Нейропедиатрия. 1993; 24: 60-7.
ИНТЕРНЕТ
Thorburn DR, Rahman S. Mitochondrial DNA-Associated Leigh Syndrome and NARP. 30 октября 2003 г. [Обновлено 17 апреля 2014 г.]. В: Pagon RA, Adam MP, Ardinger HH, et al., Редакторы. GeneReviews® [Интернет]. Сиэтл (Вашингтон): Вашингтонский университет, Сиэтл; 1993-2016 гг.Доступно по адресу: http://www.ncbi.nlm.nih.gov/books/NBK1173/ По состоянию на 16 марта 2016 г.
Онлайн-Менделирующее наследование в человеке (OMIM). Университет Джона Хопкинса. Синдром Ли; LS. Запись №: 256000. Последнее редактирование 20.01.16. Доступно по адресу: http://omim.org/entry/256000 По состоянию на 16 марта 2016 г.
Последнее редактирование 20.01.16. Доступно по адресу: http://omim.org/entry/256000 По состоянию на 16 марта 2016 г.
Страница информации о болезни Ли. Национальный институт неврологических расстройств и инсульта (NINDS). http://www.ninds.nih.gov/disorders/leighsdisease/leighsdisease.htm Последнее обновление: 16 декабря 2011 г.По состоянию на 16 марта 2016 г.
Интернет-Менделирующее наследование в человеке (OMIM). Университет Джона Хопкинса. Некротическая энцефаломиелопатия, подострая, Лиа, взрослая. Запись №: 161700. Последняя редакция 13 октября 2011 г. Доступно по адресу http://omim.org/entry/161700 Доступно с 16 марта 2016 г.
Менделирующее наследование в Интернете в Интернете (OMIM). Университет Джона Хопкинса. Дефицит пируватдегидрогеназы E1-альфа; ПДХАД. Запись №: 312170 .. 03.11.2014. Доступно по адресу: http://omim.org/entry/312170 Доступно с 16 марта 2016 г.
Болезнь Ломбеса А. Ли. Энциклопедия Orphanet. http://www.orpha.net/consor/cgi-bin/OC_Exp. php?Lng=GB&Expert=506 Последнее обновление июль 2006 г. По состоянию на 16 марта 2016 г.
php?Lng=GB&Expert=506 Последнее обновление июль 2006 г. По состоянию на 16 марта 2016 г.
Информационная страница о болезни Ли | Национальный институт неврологических расстройств и инсульта
Определение
Лечение
Прогноз
Клинические испытания
Организации
Публикации
Определение
Болезнь Ли — редкое наследственное нейрометаболическое заболевание, поражающее центральную нервную систему. Это прогрессирующее заболевание начинается у младенцев в возрасте от трех месяцев до двух лет. Реже встречается у подростков и взрослых. Болезнь Ли может быть вызвана мутациями в митохондриальной ДНК или недостатком фермента, называемого пируватдегидрогеназой. Симптомы болезни Ли обычно быстро прогрессируют. Самыми ранними признаками могут быть плохая сосательная способность, потеря контроля над головой и моторики. Эти симптомы могут сопровождаться потерей аппетита, рвотой, раздражительностью, постоянным плачем и судорогами.По мере прогрессирования заболевания симптомы могут также включать общую слабость, отсутствие мышечного тонуса и эпизоды лактоацидоза, что может привести к нарушению функции дыхания и почек.
Это прогрессирующее заболевание начинается у младенцев в возрасте от трех месяцев до двух лет. Реже встречается у подростков и взрослых. Болезнь Ли может быть вызвана мутациями в митохондриальной ДНК или недостатком фермента, называемого пируватдегидрогеназой. Симптомы болезни Ли обычно быстро прогрессируют. Самыми ранними признаками могут быть плохая сосательная способность, потеря контроля над головой и моторики. Эти симптомы могут сопровождаться потерей аппетита, рвотой, раздражительностью, постоянным плачем и судорогами.По мере прогрессирования заболевания симптомы могут также включать общую слабость, отсутствие мышечного тонуса и эпизоды лактоацидоза, что может привести к нарушению функции дыхания и почек.
При болезни Ли генетические мутации в митохондриальной ДНК мешают источникам энергии, которые управляют клетками в той области мозга, которая играет роль в двигательных движениях. Основная функция митохондрий — преобразовывать энергию глюкозы и жирных кислот в вещество, называемое аденозинтрифосфатом (АТФ). Энергия АТФ управляет практически всеми метаболическими функциями клетки. Следовательно, генетические мутации в митохондриальной ДНК приводят к хронической нехватке энергии в этих клетках, что, в свою очередь, влияет на центральную нервную систему и вызывает прогрессирующую дегенерацию двигательных функций.
Энергия АТФ управляет практически всеми метаболическими функциями клетки. Следовательно, генетические мутации в митохондриальной ДНК приводят к хронической нехватке энергии в этих клетках, что, в свою очередь, влияет на центральную нервную систему и вызывает прогрессирующую дегенерацию двигательных функций.
Существует также форма болезни Ли (называемая Х-сцепленной болезнью Ли), которая является результатом мутации в гене, который продуцирует другую группу веществ, которые важны для клеточного метаболизма. Этот ген находится только на Х-хромосоме.
×
Определение
Болезнь Ли — редкое наследственное нейрометаболическое заболевание, поражающее центральную нервную систему. Это прогрессирующее заболевание начинается у младенцев в возрасте от трех месяцев до двух лет. Реже встречается у подростков и взрослых. Болезнь Ли может быть вызвана мутациями в митохондриальной ДНК или недостатком фермента, называемого пируватдегидрогеназой.Симптомы болезни Ли обычно быстро прогрессируют. Самыми ранними признаками могут быть плохая сосательная способность, потеря контроля над головой и моторики. Эти симптомы могут сопровождаться потерей аппетита, рвотой, раздражительностью, постоянным плачем и судорогами. По мере прогрессирования заболевания симптомы могут также включать общую слабость, отсутствие мышечного тонуса и эпизоды лактоацидоза, что может привести к нарушению функции дыхания и почек.
Болезнь Ли может быть вызвана мутациями в митохондриальной ДНК или недостатком фермента, называемого пируватдегидрогеназой.Симптомы болезни Ли обычно быстро прогрессируют. Самыми ранними признаками могут быть плохая сосательная способность, потеря контроля над головой и моторики. Эти симптомы могут сопровождаться потерей аппетита, рвотой, раздражительностью, постоянным плачем и судорогами. По мере прогрессирования заболевания симптомы могут также включать общую слабость, отсутствие мышечного тонуса и эпизоды лактоацидоза, что может привести к нарушению функции дыхания и почек.
При болезни Ли генетические мутации в митохондриальной ДНК мешают источникам энергии, которые управляют клетками в той области мозга, которая играет роль в двигательных движениях.Основная функция митохондрий — преобразовывать энергию глюкозы и жирных кислот в вещество, называемое аденозинтрифосфатом (АТФ). Энергия АТФ управляет практически всеми метаболическими функциями клетки. Следовательно, генетические мутации в митохондриальной ДНК приводят к хронической нехватке энергии в этих клетках, что, в свою очередь, влияет на центральную нервную систему и вызывает прогрессирующую дегенерацию двигательных функций.
Существует также форма болезни Ли (называемая Х-сцепленной болезнью Ли), которая является результатом мутации в гене, который продуцирует другую группу веществ, которые важны для клеточного метаболизма.Этот ген находится только на Х-хромосоме.
Лечение
Наиболее распространенным средством лечения болезни Ли является тиамин или витамин B1. Пероральный бикарбонат натрия или цитрат натрия также может быть назначен для лечения лактоацидоза. В настоящее время исследователи тестируют дихлорацетат, чтобы установить его эффективность при лечении лактоацидоза.Людям с Х-сцепленной формой болезни Ли может быть рекомендована диета с высоким содержанием жиров и низким содержанием углеводов.
×
Лечение
Наиболее распространенным средством лечения болезни Ли является тиамин или витамин B1. Пероральный бикарбонат натрия или цитрат натрия также может быть назначен для лечения лактоацидоза.В настоящее время исследователи тестируют дихлорацетат, чтобы установить его эффективность при лечении лактоацидоза. Людям с Х-сцепленной формой болезни Ли может быть рекомендована диета с высоким содержанием жиров и низким содержанием углеводов.
Пероральный бикарбонат натрия или цитрат натрия также может быть назначен для лечения лактоацидоза.В настоящее время исследователи тестируют дихлорацетат, чтобы установить его эффективность при лечении лактоацидоза. Людям с Х-сцепленной формой болезни Ли может быть рекомендована диета с высоким содержанием жиров и низким содержанием углеводов.
Определение
Болезнь Ли — редкое наследственное нейрометаболическое заболевание, поражающее центральную нервную систему.Это прогрессирующее заболевание начинается у младенцев в возрасте от трех месяцев до двух лет. Реже встречается у подростков и взрослых. Болезнь Ли может быть вызвана мутациями в митохондриальной ДНК или недостатком фермента, называемого пируватдегидрогеназой. Симптомы болезни Ли обычно быстро прогрессируют. Самыми ранними признаками могут быть плохая сосательная способность, потеря контроля над головой и моторики. Эти симптомы могут сопровождаться потерей аппетита, рвотой, раздражительностью, постоянным плачем и судорогами.По мере прогрессирования заболевания симптомы могут также включать общую слабость, отсутствие мышечного тонуса и эпизоды лактоацидоза, что может привести к нарушению функции дыхания и почек.
Эти симптомы могут сопровождаться потерей аппетита, рвотой, раздражительностью, постоянным плачем и судорогами.По мере прогрессирования заболевания симптомы могут также включать общую слабость, отсутствие мышечного тонуса и эпизоды лактоацидоза, что может привести к нарушению функции дыхания и почек.
При болезни Ли генетические мутации в митохондриальной ДНК мешают источникам энергии, которые управляют клетками в той области мозга, которая играет роль в двигательных движениях. Основная функция митохондрий — преобразовывать энергию глюкозы и жирных кислот в вещество, называемое аденозинтрифосфатом (АТФ).Энергия АТФ управляет практически всеми метаболическими функциями клетки. Следовательно, генетические мутации в митохондриальной ДНК приводят к хронической нехватке энергии в этих клетках, что, в свою очередь, влияет на центральную нервную систему и вызывает прогрессирующую дегенерацию двигательных функций.
Существует также форма болезни Ли (называемая Х-сцепленной болезнью Ли), которая является результатом мутации в гене, который продуцирует другую группу веществ, которые важны для клеточного метаболизма. Этот ген находится только на Х-хромосоме.
Этот ген находится только на Х-хромосоме.
Лечение
Наиболее распространенным средством лечения болезни Ли является тиамин или витамин B1. Пероральный бикарбонат натрия или цитрат натрия также может быть назначен для лечения лактоацидоза. В настоящее время исследователи тестируют дихлорацетат, чтобы установить его эффективность при лечении лактоацидоза. Людям с Х-сцепленной формой болезни Ли может быть рекомендована диета с высоким содержанием жиров и низким содержанием углеводов.
Прогноз
Прогноз для людей с болезнью Ли плохой. Лица, у которых отсутствует активность митохондриального комплекса IV, и люди с дефицитом пируватдегидрогеназы, как правило, имеют худший прогноз и умирают в течение нескольких лет. Те, у кого есть частичный дефицит, имеют лучший прогноз и могут дожить до 6 или 7 лет. Некоторые дожили до подросткового возраста.
Некоторые дожили до подросткового возраста.
х
Прогноз
Прогноз для людей с болезнью Ли плохой. Лица, у которых отсутствует активность митохондриального комплекса IV, и люди с дефицитом пируватдегидрогеназы, как правило, имеют худший прогноз и умирают в течение нескольких лет.Те, у кого есть частичный дефицит, имеют лучший прогноз и могут дожить до 6 или 7 лет. Некоторые дожили до подросткового возраста.
Прогноз
Прогноз для людей с болезнью Ли плохой. Лица, у которых отсутствует активность митохондриального комплекса IV, и люди с дефицитом пируватдегидрогеназы, как правило, имеют худший прогноз и умирают в течение нескольких лет.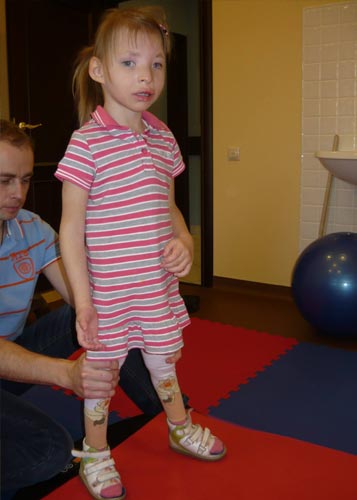 Те, у кого есть частичный дефицит, имеют лучший прогноз и могут дожить до 6 или 7 лет. Некоторые дожили до подросткового возраста.
Те, у кого есть частичный дефицит, имеют лучший прогноз и могут дожить до 6 или 7 лет. Некоторые дожили до подросткового возраста.
Определение
Болезнь Ли — редкое наследственное нейрометаболическое заболевание, поражающее центральную нервную систему. Это прогрессирующее заболевание начинается у младенцев в возрасте от трех месяцев до двух лет. Реже встречается у подростков и взрослых.Болезнь Ли может быть вызвана мутациями в митохондриальной ДНК или недостатком фермента, называемого пируватдегидрогеназой. Симптомы болезни Ли обычно быстро прогрессируют. Самыми ранними признаками могут быть плохая сосательная способность, потеря контроля над головой и моторики. Эти симптомы могут сопровождаться потерей аппетита, рвотой, раздражительностью, постоянным плачем и судорогами. По мере прогрессирования заболевания симптомы могут также включать общую слабость, отсутствие мышечного тонуса и эпизоды лактоацидоза, что может привести к нарушению функции дыхания и почек.
При болезни Ли генетические мутации в митохондриальной ДНК мешают источникам энергии, которые управляют клетками в той области мозга, которая играет роль в двигательных движениях. Основная функция митохондрий — преобразовывать энергию глюкозы и жирных кислот в вещество, называемое аденозинтрифосфатом (АТФ). Энергия АТФ управляет практически всеми метаболическими функциями клетки. Следовательно, генетические мутации в митохондриальной ДНК приводят к хронической нехватке энергии в этих клетках, что, в свою очередь, влияет на центральную нервную систему и вызывает прогрессирующую дегенерацию двигательных функций.
Существует также форма болезни Ли (называемая Х-сцепленной болезнью Ли), которая является результатом мутации в гене, который продуцирует другую группу веществ, которые важны для клеточного метаболизма. Этот ген находится только на Х-хромосоме.
Лечение
Наиболее распространенным средством лечения болезни Ли является тиамин или витамин B1. Пероральный бикарбонат натрия или цитрат натрия также может быть назначен для лечения лактоацидоза.В настоящее время исследователи тестируют дихлорацетат, чтобы установить его эффективность при лечении лактоацидоза. Людям с Х-сцепленной формой болезни Ли может быть рекомендована диета с высоким содержанием жиров и низким содержанием углеводов.
Пероральный бикарбонат натрия или цитрат натрия также может быть назначен для лечения лактоацидоза.В настоящее время исследователи тестируют дихлорацетат, чтобы установить его эффективность при лечении лактоацидоза. Людям с Х-сцепленной формой болезни Ли может быть рекомендована диета с высоким содержанием жиров и низким содержанием углеводов.
Прогноз
Прогноз для людей с болезнью Ли плохой. Лица, у которых отсутствует активность митохондриального комплекса IV, и люди с дефицитом пируватдегидрогеназы, как правило, имеют худший прогноз и умирают в течение нескольких лет.Те, у кого есть частичный дефицит, имеют лучший прогноз и могут дожить до 6 или 7 лет. Некоторые дожили до подросткового возраста.
Какие исследования проводятся?
NINDS поддерживает и поощряет широкий спектр фундаментальных и клинических исследований нейрогенетических заболеваний, таких как болезнь Ли. Цель этого исследования — понять, что вызывает эти расстройства, а затем применить эти результаты к новым способам их диагностики, лечения и профилактики.
Цель этого исследования — понять, что вызывает эти расстройства, а затем применить эти результаты к новым способам их диагностики, лечения и профилактики.
Реестр лиц с синдромом Ли был создан в Центре медицинских наук Техасского университета в Хьюстоне для сбора ценной информации о медицинском статусе пациентов и повышения понимания того, как болезнь прогрессирует. Реестр также будет служить ресурсом для исследователей, проводящих клинические испытания, ускоряя процесс поиска потенциальных субъектов исследования. Чтобы зарегистрироваться, посетите http://peopleagainstleighs.org/registry/.
Информация из MedlinePlus Национальной медицинской библиотеки
Дегенеративные нервные заболевания
Организации пациентов
Фонд эпилепсии
8301 Professional Place West, Suite 230
Landover
MD
Landover, MD 20785-2353
Тел . : 301-459-3700; 800-EFA-1000 (332-1000)
: 301-459-3700; 800-EFA-1000 (332-1000)
MitoAction
стр.О. Box 310
Нови
MI
Нови, MI 48376
Тел .: 888-648-6228
Национальная организация по редким заболеваниям (NORD)
55 Kenosia Avenue
Данбери
CT
Данбери, Коннектикут 06810-1968
Тел .: 203-744-0100; 800-999-6673; 844-259-7178 испанский
Объединенный фонд митохондриальных болезней
8085 Saltsburg Road
Люкс 201
Питтсбург
PA
Питтсбург, Пенсильвания 15239
Тел .: 412-793-8077; 888-317-УМДФ (8633)
Организации пациентов
Фонд эпилепсии
8301 Professional Place West, Suite 230
Landover
MD
Landover, MD 20785-2353
Тел . : 301-459-3700; 800-EFA-1000 (332-1000)
: 301-459-3700; 800-EFA-1000 (332-1000)
MitoAction
стр.О. Box 310
Нови
MI
Нови, MI 48376
Тел .: 888-648-6228
Национальная организация по редким заболеваниям (NORD)
55 Kenosia Avenue
Данбери
CT
Данбери, Коннектикут 06810-1968
Тел .: 203-744-0100; 800-999-6673; 844-259-7178 испанский
Объединенный фонд митохондриальных болезней
8085 Saltsburg Road
Люкс 201
Питтсбург
PA
Питтсбург, Пенсильвания 15239
Тел .: 412-793-8077; 888-317-УМДФ (8633)
Дата последнего изменения: среда, 27.03.2019 16:20
Болезнь или синдром Ли | UMDF
Полное название: Подострая некротическая энцефаломиелопатия
Симптомы: Судороги, гипотония, утомляемость, нистагм, плохие рефлексы, трудности с приемом пищи и глотания, проблемы с дыханием, плохая двигательная функция, атаксия
Болезнь Ли — прогрессирующее нейрометаболическое заболевание, которое обычно начинается в младенчестве или детстве, часто после вирусной инфекции, но также может возникать у подростков и взрослых. Он характеризуется на МРТ видимыми некротическими (мертвые или умирающие ткани) поражениями головного мозга, особенно в среднем мозге и стволе мозга.
Он характеризуется на МРТ видимыми некротическими (мертвые или умирающие ткани) поражениями головного мозга, особенно в среднем мозге и стволе мозга.
Ребенок часто выглядит нормальным при рождении, но обычно симптомы начинают проявляться в возрасте от нескольких месяцев до двух лет, хотя время может наступить намного раньше или позже. Первоначальные симптомы могут включать потерю основных навыков, таких как сосание, контроль головы, ходьба и разговор. Они могут сопровождаться другими проблемами, такими как раздражительность, потеря аппетита, рвота и судороги.Возможны периоды резкого спада или временного восстановления некоторых функций. В конце концов, у ребенка могут появиться проблемы с сердцем, почками, зрением и дыханием.
Болезнь Ли вызывается несколькими дефектами. По словам доктора Дэвида Торберна, было выявлено не менее 26 дефектов. К ним относятся дефицит пируватдегидрогеназы (PDHC) и дефекты ферментов дыхательной цепи — Комплексы I, II, IV и V. В зависимости от дефекта тип наследования может быть X-связанным доминантным (дефект на X-хромосоме и заболевание обычно встречается только у мужчин), аутосомно-рецессивный (наследуется от генов как от матери, так и от отца) и материнский (только от матери). Также могут быть спонтанные случаи, которые вообще не передаются по наследству.
Также могут быть спонтанные случаи, которые вообще не передаются по наследству.
Согласно одной из оценок заболеваемости болезнью Ли (Синдром Ли: клинические признаки и биохимические аномалии и отклонения ДНК, доктор Дэвид Торберн, доктор философии из Мельбурна, Австралия), это один на 77000 рождений или один на 40000 рождений для болезни Ли и подобной болезни Ли ( более легкая версия синдрома, часто не подтвержденная визуализацией или вскрытием). Однако это может быть заниженной оценкой, поскольку митохондриальные заболевания, как правило, недооцениваются и неправильно диагностируются.
Синдром Ли | Справочная статья по радиологии
Синдром Ли , также известный как подострая некротическая энцефаломиелопатия (SNEM) , представляет собой митохондриальное заболевание с прогрессирующей нейродегенерацией, которое неизменно приводит к смерти, обычно в детстве.
Синдром Ли встречается примерно у 1 из 40 000 рождений, хотя в некоторых популяциях заболеваемость намного выше (например, в Квебеке, Канада) 9 . Пол или расовая принадлежность неизвестны. 9 .
Пол или расовая принадлежность неизвестны. 9 .
Обычно симптомы проявляются в возрасте до 2 лет, причем проявление в более позднем детстве (ювенильная форма) или во взрослом возрасте (взрослая форма) встречается редко. Симптомы включают 6,9 :
Болезнь Ли — одно из многих митохондриальных заболеваний, обусловленное широким спектром генетических мутаций как ядерной ДНК (яДНК), так и митохондриальной ДНК (мтДНК) 8,9 .
Мутации ядерной ДНК более распространены (~ 75%) и наследуются по менделевской манере, причем встречаются как аутосомно-рецессивное, так и Х-сцепленное наследование 9 .
Случаи, связанные с митохондриальной ДНК, встречаются реже (25%) и наследуются только от матери 9 .
Некоторые мутации (например, SURF1 ) особенно разрушительны 1 .
Хроническая депривация энергии приводит к таким гистологическим признакам, как 3 :
Эти результаты аналогичны тем, которые наблюдаются при инфаркте 4 .
Генетика
Тип наследования может быть аутосомно-рецессивным или Х-сцепленным.
Маркеры
Уровень лактата в спинномозговой жидкости может быть повышен.
CT
CT демонстрирует области с низкой плотностью совпадающих областей аномального сигнала T2 на МРТ (см. Ниже) 5 . Иногда в некоторых из этих областей может наблюдаться усиление контраста 5 .
МРТ
Нарушения на МРТ неоднородны и различаются в зависимости от лежащей в основе генетической аномалии 8 . Как правило, распределение имеет тенденцию быть симметричным.
- T2: характеризуется высоким уровнем сигнала, как правило, в 3 :
- T1: обычно демонстрирует пониженный сигнал в аномальных областях T2, хотя можно увидеть некоторые области гиперинтенсивности, а также некоторое улучшение
- DWI: в острых условиях может быть очевидна некоторая ограниченная диффузия
- МР-спектроскопия
- повышенный холин
- иногда повышенный лактат
- уменьшенный NAA
Лечение и прогноз
Прогноз плохой, смерть обычно наступает в детстве. Чем позже наступление, тем медленнее ухудшение. Чаще всего смерть наступает из-за дыхательной недостаточности 6 .
Чем позже наступление, тем медленнее ухудшение. Чаще всего смерть наступает из-за дыхательной недостаточности 6 .
Факторы, связанные с худшим исходом: 10 :
- Начало заболевания до 6 месяцев
- поступление в реанимацию
- Поражения ствола головного мозга
- Пик лактата MRS
История и этимология
Назван в честь Арчибальда Дени Ли , Британский невропатолог, впервые описавший состояние в 1951 году 2,9 .
Синдром беспокойных ног — Симптомы и причины
Обзор
Синдром беспокойных ног (СБН) — это состояние, которое вызывает неконтролируемое желание пошевелить ногами, обычно из-за неприятных ощущений. Обычно это происходит в вечерние или ночные часы, когда вы сидите или лежите. Движение временно облегчает неприятные ощущения.
Синдром беспокойных ног, также известный как болезнь Уиллиса-Экбома, может начаться в любом возрасте и обычно ухудшается с возрастом. Это может нарушить сон, что мешает повседневной деятельности.
Это может нарушить сон, что мешает повседневной деятельности.
Простые шаги по уходу за собой и изменение образа жизни могут помочь облегчить симптомы. Лекарства также помогают многим людям с RLS .
Продукты и услуги
Показать больше товаров от Mayo ClinicСимптомы
Главный симптом — позывы пошевелить ногами. Общие сопроводительные характеристики RLS включают:
- Ощущения, начинающиеся после отдыха. Ощущение обычно начинается после того, как вы лежите или сидите в течение длительного времени, например, в машине, самолете или кинотеатре.
- Рельеф с движением. Ощущение RLS ослабевает при движении, например при растяжении, покачивании ногами, при ходьбе или ходьбе.
- Обострение симптомов вечером. Симптомы возникают в основном ночью.
- Подергивание ног в ночное время. RLS может быть связано с другим, более распространенным состоянием, называемым периодическим движением конечностей во сне, которое заставляет ваши ноги подергиваться и пинаться, возможно, в течение всей ночи, пока вы спите.
 Симптомы возникают в основном ночью.
Симптомы возникают в основном ночью.Люди обычно описывают симптомы RLS как ненормальные, неприятные ощущения в ногах или ступнях. Обычно они возникают с обеих сторон тела. Реже ощущения поражают руки.
Ощущения, которые обычно возникают в конечностях, а не на коже, описываются как:
- Ползание
- Ползучая
- Тянуть
- Пульсирующий
- Болит
- Зуд
- Электрический
Иногда ощущения сложно объяснить.Люди с RLS обычно не описывают это состояние как мышечную судорогу или онемение. Однако они постоянно описывают желание пошевелить ногами.
Однако они постоянно описывают желание пошевелить ногами.
Обычно симптомы колеблются по степени тяжести. Иногда симптомы исчезают на какое-то время, а затем возвращаются.
Когда обращаться к врачу
Некоторые люди с RLS никогда не обращаются за медицинской помощью, потому что боятся, что их не воспримут всерьез. Но RLS может мешать вашему сну, вызывать дневную сонливость и влиять на качество вашей жизни.Поговорите со своим врачом, если вы думаете, что у вас может быть RLS .
Причины
Часто причина RLS неизвестна. Исследователи подозревают, что это состояние может быть вызвано дисбалансом химического вещества дофамина в мозге, который посылает сигналы для контроля движения мышц.
Наследственность
Иногда RLS передается в семье, особенно если заболевание начинается до 40 лет.Исследователи определили участки хромосом, где могут присутствовать гены RLS .
Беременность
Беременность или гормональные изменения могут временно ухудшить RLS признаки и симптомы. Некоторые женщины получают RLS впервые во время беременности, особенно в последнем триместре. Однако обычно симптомы исчезают после родов.
Факторы риска
RLS может развиться в любом возрасте, даже в детстве.Расстройство чаще встречается с возрастом и чаще встречается у женщин, чем у мужчин.
RLS обычно не связано с серьезной основной медицинской проблемой. Однако иногда он сопровождает другие условия, например:
Однако иногда он сопровождает другие условия, например:
- Периферическая невропатия. Это повреждение нервов рук и ног иногда происходит из-за хронических заболеваний, таких как диабет и алкоголизм.
- Дефицит железа. Даже без анемии дефицит железа может вызвать или усугубить RLS .Если у вас в анамнезе были кровотечения из желудка или кишечника, у вас были обильные менструальные периоды или вы неоднократно сдали кровь, у вас может быть дефицит железа.
- Почечная недостаточность. Если у вас почечная недостаточность, у вас также может быть дефицит железа, часто с анемией. Когда почки не функционируют должным образом, запасы железа в крови могут уменьшаться. Это и другие изменения химического состава тела могут вызвать или ухудшить состояние RLS .
- Заболевания спинного мозга. Поражения спинного мозга в результате повреждения или травмы были связаны с RLS . Наличие анестезии спинного мозга, такой как спинальная блокада, также увеличивает риск развития RLS .
 Наличие анестезии спинного мозга, такой как спинальная блокада, также увеличивает риск развития RLS .
Наличие анестезии спинного мозга, такой как спинальная блокада, также увеличивает риск развития RLS .Осложнения
Хотя RLS не приводит к другим серьезным заболеваниям, симптомы могут варьироваться от едва беспокоящих до выводящих из строя. Многие люди с RLS испытывают трудности с засыпанием или засыпанием.
Тяжелая RLS может вызвать заметное ухудшение качества жизни и привести к депрессии. Бессонница может привести к чрезмерной дневной сонливости, но RLS может мешать дремоте.
21 января 2020 г.
Синдром беспокойных ног: диагностика и тестирование
Если вы испытываете боль в ногах по ночам, вам может быть интересно, нет ли у вас синдрома беспокойных ног (СБН). Боль в ногах в ночное время может быть вызвана множеством состояний, включая RLS. Однако это также может произойти, если вы слишком много времени проводите сидя в течение дня или спите, ограничивая приток крови к нижним конечностям.Боль в ногах может быть обычным явлением во время беременности или быть побочным эффектом приема лекарств. Боль в ногах также может быть вызвана хроническим заболеванием, например артритом, заболеванием периферических артерий или диабетом.
Боль в ногах в ночное время может быть вызвана множеством состояний, включая RLS. Однако это также может произойти, если вы слишком много времени проводите сидя в течение дня или спите, ограничивая приток крови к нижним конечностям.Боль в ногах может быть обычным явлением во время беременности или быть побочным эффектом приема лекарств. Боль в ногах также может быть вызвана хроническим заболеванием, например артритом, заболеванием периферических артерий или диабетом.
Как узнать, что за болью в ноге стоит СБН, а не что-то еще? Ниже мы объясняем симптомы, которые отделяют RLS от других состояний, связанных с болью в ногах, и того, чего вы можете ожидать во время диагностического процесса.
Что такое синдром беспокойных ног?
Синдром беспокойных ног, также известный как болезнь Уиллиса-Экбома, описывается как неврологическое сенсорное расстройство и расстройство движений во сне, характеризующееся дискомфортными ощущениями в ногах, обычно в положении лежа, и непреодолимым желанием пошевелить ими, чтобы испытать облегчение. .
.
Как узнать, есть ли у вас СБН? У вас могут возникнуть следующие симптомы:
- У вас непреодолимое желание двигаться или растягиваться, часто из-за неприятных ощущений в ногах. Эти ощущения отличаются от онемения или судорог, связанных с чарлиской лошадью. Скорее пациенты с RLS описывают их как подергивания, зуд, боль, ползание, покалывание или дергание. Симптомы варьируются от неприятных до болезненных. Синдром беспокойных ног называется так, потому что ощущения в первую очередь ощущаются в ногах, хотя до 57 процентов людей могут испытывать аналогичные ощущения в руках.Ощущения обычно затрагивают обе ноги, но могут проявляться только на одной ноге или чередоваться между ногами.
- Частичное или временное движение снимает эти ощущения. Лица с СБН могут почувствовать облегчение от ударов ногами, трения, ходьбы, ходьбы или передвижения. Как только вы перестанете двигаться, ощущения могут повториться.
- Симптомы начинаются или усиливаются, когда вы малоподвижны, особенно когда вы лежите, сидите или отдыхаете. Например, вы можете лежать в постели, отдыхать на диване или сидеть в самолете.
- Симптомы в основном возникают ночью или усиливаются вечером и ночью . Обычно утром симптомы практически отсутствуют, а ночью симптомы усиливаются.
- Ваши симптомы не вызваны другим медицинским или поведенческим заболеванием , таким как артрит, судороги ног или привычное постукивание ногой.
- Ваши симптомы нарушают ваш сон, вызывают у вас стресс или иным образом ухудшают ваше самочувствие или способность нормально функционировать. Плохой сон — основная причина, по которой люди обращаются за помощью по поводу своих симптомов RLS, и затрагивает от 60 до 90 процентов людей с RLS.В свою очередь, плохой сон может иметь негативные психические, физические или поведенческие последствия, которые затрудняют преодоление СБН.
 Например, вы можете лежать в постели, отдыхать на диване или сидеть в самолете.
Например, вы можете лежать в постели, отдыхать на диване или сидеть в самолете.Если вы столкнулись с этими симптомами, обратитесь к врачу.
Есть ли тест на синдром беспокойных ног?
Не существует специального диагностического теста для синдрома беспокойных ног.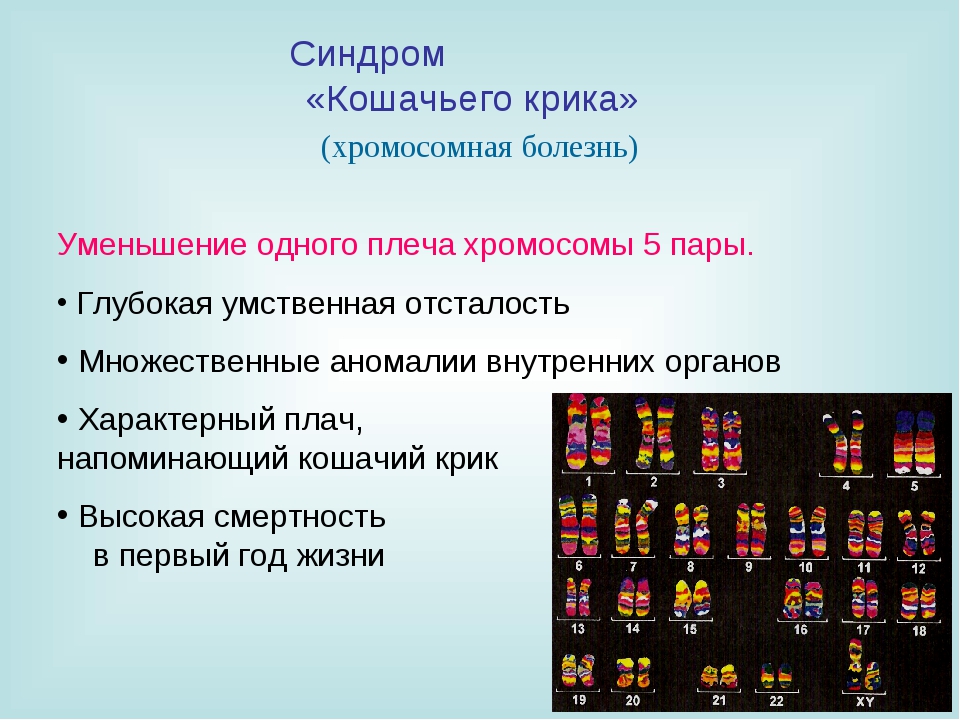 Если вы считаете, что у вас может быть СБН, вам следует записаться на прием к врачу. Они проведут тщательный сбор анамнеза и проведут физический осмотр, чтобы определить, что вызывает ваши симптомы.Могут потребоваться другие тесты и исследования, чтобы исключить медицинские условия, проявляющиеся схожими симптомами. А пока вы можете отслеживать симптомы СБН дома с помощью дневника сна.
Если вы считаете, что у вас может быть СБН, вам следует записаться на прием к врачу. Они проведут тщательный сбор анамнеза и проведут физический осмотр, чтобы определить, что вызывает ваши симптомы.Могут потребоваться другие тесты и исследования, чтобы исключить медицинские условия, проявляющиеся схожими симптомами. А пока вы можете отслеживать симптомы СБН дома с помощью дневника сна.
Как выполнить самотестирование для RLS
Найдите записную книжку или используйте приложение для заметок на телефоне, чтобы использовать его как дневник сна. Каждую ночь, когда вы ложитесь спать, и каждое утро, когда вы просыпаетесь, отвечайте на следующие вопросы. Вы будете использовать эту информацию, чтобы ответить на вопросы, которые врач задаст вам во время приема.
Вопросы о качестве сна:
- В какое время вы заснули? Вам потребовалось больше времени, чем обычно, чтобы заснуть из-за симптомов СБН?
- Во сколько вы проснулись? Вы проснулись естественным путем или по будильнику?
- Сколько всего времени вы провели во сне?
- Вы вообще просыпались ночью? Запишите, сколько раз вы просыпались, как долго и что заставляло вас просыпаться, если применимо (например, потребность в мочеиспускании или кошмар).
- Вы вообще спали днем? Сколько раз и как долго?

Вопросы по RLS:
- Какие симптомы RLS вы испытываете? Запишите, что они чувствуют.
- В какое время появились симптомы и что вы делали?
- Где вы почувствовали симптомы (например, в ногах или руках)?
- Насколько серьезными были симптомы?
- Как долго длились симптомы?
- Что помогло облегчить ваши симптомы?
Вопросы по образу жизни:
- Какие упражнения вы выполняли каждый день?
- Вы принимали лекарства? Перечислите их вместе с дозой.
- Были ли у вас кофеин, алкоголь или никотин?
- Как вы себя чувствовали днем? Ментально, физически и эмоционально?
В течение нескольких недель вы можете заметить определенные тенденции. Например, ваши симптомы могут ухудшиться, когда вы пьете кофеин. Обратите внимание на эти тенденции и поделитесь ими со своим врачом.
Как врач ставит диагноз RLS
Как правило, врач должен соответствовать следующим диагностическим критериям, чтобы диагностировать RLS:
- Позыв к движению ногами, обычно сопровождающийся или вызываемый дискомфортными и неприятными ощущениями в ногах
- Они должны начинаться или ухудшаться в периоды отдыха
- Быть частично или полностью освобожденным от движения
- Возникают исключительно или преимущественно вечером или ночью, а не днем
- Вышеуказанные признаки не учитываются исключительно как симптомы другого медицинского или поведенческого состояния
- Симптомы RLS вызывают беспокойство, дистресс, нарушение сна или ухудшение умственной, физической, эмоциональной, социальной, профессиональной, образовательной, поведенческой или других важных сфер жизнедеятельности.

Проблема диагностики RLS заключается, по крайней мере, частично, в том, чтобы полагаться на сообщение о субъективных симптомах, что делает использование дневника сна критически важным для документирования своего опыта.
Когда вы встретитесь со своим врачом, он проведет начальное диагностическое интервью для RLS. Они могут задать вам контрольные вопросы, подобные приведенным ниже:
- Испытывали ли вы за последнюю неделю неприятные ощущения в ногах, сопровождаемые непреодолимым желанием пошевелить ими, чтобы почувствовать облегчение?
- Как бы вы описали ощущения, которые испытываете в ногах?
- Ваши симптомы начинаются или ухудшаются, когда вы отдыхаете, сидите или лежите?
- Ваши симптомы проявляются ночью или усиливаются к вечеру?
- Есть ли у вас трудности с засыпанием или засыпанием из-за ваших симптомов?
- Ваши симптомы облегчаются от движения?
- Есть ли у вас какие-либо заболевания?
- Вы принимаете какие-либо лекарства?
- Есть ли у кого-нибудь в вашей семье СБН?
- Какая у вас типичная диета и какой режим упражнений?
- Возможно ли у вас дефицит железа?
- Вы беременны?
Ваш врач задаст эти вопросы, чтобы исключить другие потенциальные причины ваших симптомов RLS, такие как беременность, дефицит железа или терминальная почечная недостаточность. Беременные женщины в два-три раза чаще сообщают о симптомах СБН, особенно в третьем триместре, а люди с хронической почечной недостаточностью в два-пять раз чаще. Семейный анамнез RLS — еще один фактор риска RLS. Употребление кофеина, алкоголя и никотина также может усугубить симптомы, как и некоторые лекарства, в том числе лекарства от тошноты, нейролептики и антидепрессанты.
Беременные женщины в два-три раза чаще сообщают о симптомах СБН, особенно в третьем триместре, а люди с хронической почечной недостаточностью в два-пять раз чаще. Семейный анамнез RLS — еще один фактор риска RLS. Употребление кофеина, алкоголя и никотина также может усугубить симптомы, как и некоторые лекарства, в том числе лекарства от тошноты, нейролептики и антидепрессанты.
На основании ваших ответов ваш врач может назначить анализ крови или направить вас к специалисту по сну.Анализ крови поможет исключить дефицит железа, фактор риска СБН. Исследование ночного сна, известное как полисомнограмма, может быть назначено, если ваш врач считает, что ваши симптомы могут быть связаны с другим расстройством сна, таким как расстройство периодических движений конечностей или апноэ во сне. Эти расстройства могут сочетаться с СБН или ухудшать симптомы.
В зависимости от вашего диагноза ваш врач разработает для вас план лечения. Например, прием добавок железа, отпускаемых без рецепта, может помочь устранить основной дефицит железа. Некоторые легкие или умеренные симптомы СБН можно облегчить с помощью изменения образа жизни, например уменьшения потребления алкоголя и никотина или улучшения гигиены сна. Другие домашние средства могут быть полезны для уменьшения симптомов, включая массаж ног, теплые ванны, RLS-обертывания для ног и горячую / холодную терапию.
Некоторые легкие или умеренные симптомы СБН можно облегчить с помощью изменения образа жизни, например уменьшения потребления алкоголя и никотина или улучшения гигиены сна. Другие домашние средства могут быть полезны для уменьшения симптомов, включая массаж ног, теплые ванны, RLS-обертывания для ног и горячую / холодную терапию.
Для многих людей RLS — это пожизненное состояние, но симптомы можно контролировать, чтобы вы могли меньше испытывать дискомфорт и больше спать.
- Была ли эта статья полезной?
- Да Нет
