
Фку болезнь – диета и питание. :: Polismed.com
причины, симптомы, диагностика и лечение

Фенилкетонурия – наследственное нарушение аминокислотного обмена, обусловленное недостаточностью печеночных ферментов, участвующих в метаболизме фенилаланина до тирозина. Ранними признаками фенилкетонурии служат рвота, вялость или гиперактивность, запах плесени от мочи и кожи, задержка психомоторного развития; типичные поздние признаки включают олигофрению, отставание в физическом развитии, судороги, экзематозные изменения кожи и др. Скрининг новорожденных на фенилкетонурию проводится еще в родильном доме; последующая диагностика включает молекулярно-генетическое тестирование, определение концентрации фенилаланина в крови, биохимический анализ мочи, ЭЭГ, МРТ головного мозга. Лечение фенилкетонурии заключается в соблюдении специальной диеты.
Общие сведения
Фенилкетонурия (болезнь Феллинга, фенилпировиноградная олигофрения) – врожденная, генетически обусловленная патология, характеризующаяся нарушением гидроксилирования фенилаланина, накоплением аминокислоты и ее метаболитов в физиологических жидкостях и тканях с последующим тяжелым поражением ЦНС. Фенилкетонурия впервые описана А. Феллингом в 1934 г.; встречается с частотой 1 случай на 10 000 новорожденных. В неонатальном периоде фенилкетонурия не имеет клинических проявлений, однако поступление фенилаланина с пищей вызывает манифестацию заболевания уже в первом полугодии жизни, а в дальнейшем приводит к тяжелым нарушениям развития ребенка. Именно поэтому пресимптоматическое выявление фенилкетонурии у новорожденных является важнейшей задачей неонатологии, педиатрии и генетики.

Фенилкетонурия
Причины фенилкетонурии
Фенилкетонурия является заболеванием с аутосомно-рецессивным характером наследования. Это означает, что для развития клинических признаков фенилкетонурии ребенок должен унаследовать по одной дефектной копии гена от обоих родителей, являющихся гетерозиготными носителями мутантного гена.
Чаще всего к развитию фенилкетонурии приводит мутация гена, кодирующего фермент фенилаланин-4-гидроксилазу и расположенного на длинном плече 12 хромосомы (локус12q22-q24.1). Это, так называемая, классическая фенилкетонурия I типа, составляющая 98% всех случаев заболевания. Гиперфенилаланинемия может достигать 30 мг% и выше. При отсутствии лечения данный вариант фенилкетонурии сопровождается глубокой умственной отсталостью.
Кроме классической формы, различают атипичные варианты фенилкетонурии, протекающие с той же клинической симптоматикой, но не поддающиеся коррекции диетотерапией. К ним относятся фенилкетонурия II типа (недостаточность дегидроптеринредуктазы), фенилкетонурия III типа (дефицит тетрагидробиоптерина) и другие, более редкие варианты.
Вероятность рождения ребенка, больного фенилкетонурией, повышается при заключении близкородственных браков.
Патогенез фенилкетонурии
В основе классической формы фенилкетонурии лежит недостаточность фермента фенилаланин-4-гидроксилазы, участвующего в конверсии фенилаланина в тирозин в митохондриях гепатоцитов. В свою очередь, производный тирозина – тирамин является исходным продуктом для синтеза катехоламинов (адреналина и норадреналина), а дийодтирозин – для образования тироксина. Кроме этого, результатом метаболизма фенилаланина служит образование пигмента меланина.
Наследственная недостаточность фермента фенилалаиин-4-гидроксилазы при фенилкетонурии приводит к нарушению окисления фенилаланина, поступающего с пищей, в результате чего его концентрация в крови (фенилаланинемия) и спинномозговой жидкости значительно возрастает, а уровень тирозина соответственно падает. Избыточное содержание фенилаланина устраняется путем повышенной экскреции с мочой его метаболитов — фенилпировиноградной, фенилмолочной и фенилуксусной кислот.
Нарушение обмена аминокислот сопровождается нарушением миелинизации нервных волокон, снижением образования нейромедиаторов (дофамина, серотонина и др.), запускающими патогенетические механизмы задержки умственного развития и прогредиентное слабоумие.
Симптомы фенилкетонурии
Новорожденные с фенилкетонурией не имеют клинических признаков заболевания. Обычно манифестация фенилкетонурии у детей происходит в возрасте 2-6 месяцев. С началом кормления в организм ребенка начинает поступать белок грудного молока либо его заменителей, что приводит к развитию первых, неспецифических симптомов – вялости, иногда – беспокойства и гипервозбудимости, срыгивания, мышечной дистонии, судорожного синдрома. Одним из ранних патогномоничных признаков фенилкетонурии служит упорная рвота, которая нередко ошибочно расценивается как проявление пилоростеноза.
Ко второму полугодию становится заметным отставание ребенка в психомоторном развитии. Ребенок становится менее активным, безучастным, перестает узнавать близких, не пытается садиться и вставать на ножки. Аномальный состав мочи и пота обусловливают характерный «мышиный» запах (запах плесени), исходящий от тела. Часто наблюдается шелушение кожи, дерматиты, экзема, склеродермия.
У детей с фенилкетонурией, не получающих лечения, выявляется микроцефалия, прогнатия, позднее (после 1,5 лет) прорезывание зубов, гипоплазия эмали. Отмечается задержка речевого развития, а к 3-4 годам выявляется глубокая олигофрения (идиотия) и практически полное отсутствие речи.
Дети с фенилкетонурией имеют диспластическое телосложение, нередко — врожденные пороки сердца, вегетативные дисфункции (потливость, акроцианоз, артериальную гипотонию), страдают запорами. К фенотипическим особенностям детей, страдающих фенилкетонурией, следует отнести светлую кожу, глаза и волосы. Для ребенка с фенилкетонурией характерны специфическая поза «портного» (согнутые в суставах верхние и нижние конечности), тремор рук, шаткая, семенящая походка, гиперкинезы.
Клинические проявления фенилкетонурии II типа характеризуются тяжелой степенью умственной отсталости, повышенной возбудимостью, судорогами, спастическим тетрапарезом, сухожильной гиперрефлексией. Прогрессирование заболевание может приводить к гибели ребенка в возрасте 2-З лет.
При фенилкетонури III типа развивается триада признаков: микроцефалия, олигофрения, спастический тетрапарез.
Диагностика фенилкетонурии
В настоящее время диагностика фенилкетонурии (а также галактоземии, врожденного гипотиреоза, адрено-генитального синдрома и муковисцидоза) входит в программу неонатального скрининга, осуществляемого всем новорожденным.
Скрининг-тест проводится на 3-5 день жизни доношенного и 7 день жизни недоношенного ребенка путем забора образца капиллярной крови на специальный бумажный бланк. При обнаружении гиперфенилаланемии более 2,2 мг% ребенка направляют к детскому генетику для повторного обследования.
Для подтверждения диагноза фенилкетонурии проверяется концентрация фенилаланина и тирозина в крови, определяют активность печеночных ферментов (фенилаланингидроксилазы), выполняется биохимическое исследование мочи (определение кетоновых кислот), метаболитов катехоламинов в моче и др. Дополнительно проводится ЭЭГ и МРТ головного мозга, осмотр ребенка детским неврологом.
Генетический дефект при фенилкетонурии может быть обнаружен еще на этапе беременности в ходе инвазивной пренатальной диагностики плода (хорионбиопсии, амниоцентеза, кордоцентеза).
Дифференциальный диагноз фенилкетонурии проводят с внутричерепной родовой травмой новорожденных, внутриутробными инфекциями, другими нарушениями обмена аминокислот.
Лечение фенилкетонурии
Основополагающим фактором в лечении фенилкетонурии является соблюдение диеты, ограничивающей поступление белка в организм. Лечение рекомендуется начинать при концентрации фенилаланина >6 мг%. Для грудных детей разработаны специальные смеси — Афенилак, Лофенилак; для детей старше 1 года – Тетрафен, Фенил-фри; старше 8 лет — Максамум-ХР и др. Основу диеты составляют низкобелковые продукты — фрукты, овощи, соки, белковые гидролизаты и аминокислотные смеси. Расширение диеты возможно после 18 лет в связи с возрастанием толерантности к фенилаланину. В соответствии с российским законодательством обеспечение лиц, страдающих фенилкетонурией, лечебным питанием, должна осуществляться бесплатно.
Больным назначается прием минеральных соединений, витаминов группы В и др.; по показаниям — ноотропные средства, антиконвульсанты. В комплексной терапии фенилкетонурии широко используется общий массаж, ЛФК, иглорефлексотерапия.
Дети, страдающие фенилкетонурией, находятся под наблюдением участкового педиатра и психоневролога; нередко нуждаются в помощи логопеда и дефектолога. Необходим тщательный мониторинг нервно-психического статуса детей, контроль уровня фенилаланина в крови и показателей электроэнцефалограммы.
Атипичные формы фенилкетонурии, не поддающиеся лечению диетой, требуют назначения гепатопротекторов, противосудорожных средств, заместительной терапии леводопой, 5-гидрокситриптофаном.
Прогноз и профилактика фенилкетонурии
Профилактика осложнений фенилкетонурии заключается в проведении массового скрининга новорожденных, раннего назначения и длительного соблюдения диетического питания.
С целью оценки риска рождения ребенка с фенилкетонурией предварительное генетическое консультирование должны пройти супружеские пары, уже имеющие больного ребенка, состоящие в кровнородственном браке, имеющие родственников с данным заболеванием. Женщины с фенилкетонурией, планирующие беременность, должны соблюдать строгую диету до зачатия и во время беременности для исключения повышения уровня фенилаланина и его метаболитов и нарушения развития генетически здорового плода. Риск рождения ребенка с фенилкетонурией у родителей-носителей дефектного гена, составляет 1:4.
www.krasotaimedicina.ru
что это за заболевание, фото больных, тип наследования
Природа не устаёт удивлять своей мудростью и щедростью. Одним она дарит радость иметь здоровых детей, другим готовит на этом пути серьёзные испытания. Фенилкетонурия (ФКУ) — патология, способная сломать малышу жизнь, если взрослые не примут вовремя мер по её лечению. Подобные «сюрпризы» не заставят врасплох, если родители с серьёзностью отнесутся к мерам профилактики болезни.

Что такое фенилкетонурия
Это заболевание, обусловленное генетически и связанное с нарушением превращения аминокислоты фенилаланина (ФА) в тирозин. У ФКУ аутосомно-рецессивный тип наследования. Патология проявляется, когда малыш получает по одному дефектному гену от каждого из родителей.
Болезнь впервые описана в 1934 году. Сегодня врачи спасают тысячи детей с диагнозом ФКУ от слабоумия, серьёзных нарушений в работе внутренних органов, летального исхода. Пациенты, получившие лечение, становятся адаптированными к жизни в социуме, по умственным способностям не уступают ровесникам.
У детей фенилкетонурия встречается с одинаковой частотой вне зависимости от пола. В Турции 1 больной приходится на 4 тыс. новорождённых, в Японии 1 на 80000, в России 1 на 7000. На долю классической (тяжёлой) формы ФКУ приходится подавляющая часть клинических случаев, птерин–зависимые разновидности встречаются гораздо реже (1—3%). Код по МКБ 10: E70.
Симптомы заболевания
Патология опасна тем, что её признаки трудно распознаваемы. Манифестация отмечается в 2—6 месяцев, когда в организм попадает белок, содержащий фенилаланин. Проявляется в виде следующих отклонений и нарушений:
- Частая интенсивная рвота.
- Необоснованное беспокойство.
- Вялость, слабые двигательные реакции.
- Рассеянный блуждающий взгляд.
- Отсутствие эмоций.
К полугодовалому возрасту малыш не узнаёт маму, у него задерживается психомоторика, нарушается мышечный тонус, отсутствует оживлённая реакция на окружающий мир. Со временем симптомы только нарастают. У детей старше 3 лет уже замечаются:
- Психотические расстройства.
- Расторможенность.
- Нарушения поведения.
- Быстрая утомляемость.
- Физическое и умственное отставание вплоть до идиотии.
У детей запаздывают физиологические процессы: прорезывание зубов, способность переворачиваться, сидеть. У них:
- Специфическая стойка в виде широко расставленных ног, опущенных плеч и головы.
- Походка покачивающаяся, шаги маленькие.
- Возможны непроизвольные движения, тремор с развитием эпилепсии.
- Гипергидроз, кисти рук и стопы синюшного цвета.
- Моча и пот с «мышиным» запахом из-за содержащейся в них фенилуксусной кислоты.
Без лечения не избежать экзематозных изменений на коже, фолликулярного кератоза, гнойных инфекций, возникновения судорог разной степени интенсивности. Постепенно присоединяются: гиперактивность, аномалии в работе зрительного анализатора, паркинсонизм, выполнение бесцельных движений, глубокая задержка моторики — каждый третий малыш не ходит, 60% не говорят.
Фотографии больных детей



Виды
ФКУ может быть I, II и III типа в зависимости от дефицита в обмене ФА трёх основных ферментов. По другой условной классификации, применяемой в РФ, формы болезни зависят от уровня фенилаланина в крови (мкмоль/л):
- Лёгкая: 20—600.
- Среднетяжёлая: от 600 до 900.
- Классическая: от 1200.
Для сравнения у здоровых детей этот показатель колеблется от 0 до 120.
Причины
Тяжёлая форма ФКУ связана с недостатком фенилаланин-4-монооксигеназы (ФАГ) — фермента печени, почек и поджелудочной железы, обеспечивающего превращение ФА в тирозин. Ген, кодирующий ФАГ, расположен на длинном плече 12 хромосомы. ФА и его производные накапливаются в биологических жидкостях, где тормозят активность ряда ферментов: окисляющих глюкозу, синтезирующих гормон серотонин, нейромедиаторы, пигмент меланин. В результате нарушения аминокислотного обмена развиваются тяжёлые поражения головного мозга и печени.
Аминокислота относится к группе незаменимых, то есть неспособных образовываться в организме человека соединений, а поступающих только с пищей. Минимальная суточная потребность в ней для детей до года составляет 50—60 мг/кг веса, в более старшем возрасте от 15 до 40 мг. Избыток фенилаланина токсичен, приводит к тяжёлым нарушениям психического развития. Выделяется с мочой, что и обусловило название заболевания.
Наследственность
ФКУ наследуется как рецессивный признак. Патология проявляется, если малыш получил по одному мутировавшему гену от каждого из родителей. Те могут быть фенотипически (внешне) здоровыми, но являться носителями дефектных аллелей. При этом риск рождения больного младенца составляет 25%.
Диагностика
Для ребёнка важно уже с первых дней жизни приобретать различные навыки, без которых невозможно нормальное развитие. Раннее выявление ФКУ I обеспечивает полную реабилитацию малыша, его адаптацию в окружающем мире. Благодаря методике скрининга фенилкетонурии здоровье новорождённого находится под контролем вне зависимости от региона России, где тот появился на свет. В программе задействованы все детские медучреждения, роддомы, медико-генетические центры.
Суть скрининг-теста состоит в заборе на 4—5-й день жизни младенца нескольких капель крови для анализа по выявлению ФА и тирозина. Биологическая жидкость исследуется при помощи радиоиммунного метода. Результаты в виде соответствующего штампа заносятся в обменную карту. Ошибка может возникнуть при недоношенности ребёнка. Для уточнения диагноза или его опровержения проводится повторный анализ. Менее точные результаты получаются при тестировании мочи.
При положительном итоге врач приглашает родителей для беседы и незамедленного начала лечебных мероприятий. Может понадобиться помощь медицинского генетика и эндокринолога для назначения пробы Феллинга, теста Гатри, хроматографии, флуориметрии, МРТ и ЭЭГ-исследования. При поиске дефектного гена берутся образцы крови или слюны родителей.
Для ФКУ II характерно прогрессирование болезни вне зависимости от диетотерапии. Патология сопровождается выраженной умственной отсталостью, судорогами, гиперрефлексией. К 2—3 годам возможен летальный исход. При ФКУ III наблюдается триада симптомов:
- Спастический тетрапарез.
- Микроцефалия.
- Глубокая умственная отсталость.
У каждой формы свой патогенез, клиническая картина и методы лечения. Для атипичных вариантов ФКУ (до 1% от всех случаев) оно сложное из-за устойчивости к диетотерапии. Поэтому сначала определяют тип заболевания, детально исследовав биохимию патологии.
Лечение фенилкетонурии
Своевременное лечение даёт хороший эффект. Ребёнка с ФКУ не госпитализируют. Родителям предоставляется квалифицированная консультативная помощь, малышу бесплатно выдают смеси, не содержащих животные протеины как часть специализированной диеты — единственного эффективного метода лечения ФКУ. Ребёнок получает специальные белковые гидролизаты с минимумом ФА или без него, но с достаточным уровнем витаминов и кальция.
При лечении грудное кормление ограничено (раньше полностью исключалось). Молоко сцеживают и дают согласно расчётам. Когда уровень фенилаланина нормализуется, ребёнка начинают прикармливать животным белком во избежание распада собственных тканей и истощения младенца. Включение фруктов, овощей, жиров, углеводов производится при строгом учёте уровня фенилаланина.
Сегодня не проблема найти диетический продукт или белковую смесь с определённой концентрацией ФА. По российскому законодательству дети, страдающие от ФКУ, получают бесплатное специальное питание. В РФ разрешены следующие составы:
- Афенилак.
- XP Аналог LCP.
- MD мил ФКУ-0.
Специальная диета строго соблюдается до 6 лет. Из рациона исключаются: хлеб, молочные продукты, яйца, рыба, мясо, а также Аспартам — искусственный подсластитель, который используют при изготовлении газированных напитков, конфет и жевательных резинок. Со временем нейроны становятся менее чувствительными к избытку ФА и после 16—18 лет подростки переходят на обычное питание. Но прекращение лечения может спровоцировать страх. Возвращение к диетотерапии улучшает психическое состояние пациентов.
У взрослых с фенилкетонурией наблюдаются лабильность настроения, депрессии. При высоких дозах ФА они страдают от деконцентрации и бессонницы, недостаточной мотивации, схематичности при принятии решений, импульсивности, потери способности рационально оценивать ситуации.
Лечение улучшает познавательные функции даже у страдающих тяжёлой умственной отсталостью. Если болезнь была поздно диагностирована, то соблюдение строгого режима питания позволяет снизить агрессивность, улучшить контакт пациентов с окружением. В некоторых случаях требуется пожизненное соблюдение диеты.
Лечение контролируют психиатр и медицинский генетик. При необходимости может понадобиться помощь логопеда. Больший эффект достигается при использовании ЛФК, массажа, иглорефлексотерапии.
Профилактика и прогноз
Устранить дефектный признак невозможно, поэтому своевременная диетотерапия помогает предотвратить развитие тяжёлых последствий. При планировании беременности генетический анализ назначается, если у родственников были случаи рождения детей с фенилкетонурией.
При своевременном выявлении и лечении у ФКУ благоприятный долгосрочный прогноз. Малыш в целом развивается нормально. Без диеты инвалидность наступает уже в первый год жизни. Соблюдение особого режима питания помогает предотвратить судорожные припадки и умственную неполноценность детей. Диета показана и будущим матерям с диагнозом «фенилкетонурия» во избежание микроцефалии, врождённых пороков сердца, слабоумия у ребёнка.
Сегодня разрабатываются и используются новые альтернативные методы лечения ФКУ, например, препаратом Сапроптерин для снижения уровня фенилаланина. Он эффективен при лёгких формах патологии. В России такие лекарства пока не применяются.
Елена Малышева в передаче Жить Здорово
Заключение врача
Фенилкетонурия — опасная болезнь, но её можно победить. В этой борьбе многое зависит от родителей. Им приходится учиться мотивировать ребёнка к соблюдению диеты, а не злоупотреблять одними запретами. Если контролировать уровень ФА, избегать запрещённых продуктов, то особенный ребёнок сможет развиваться, реализуя всё то, что заложено в нём природой.
wiki-farm.ru
Фенилкетонурия — виды, симптомы, лечение
Краткая характеристика заболевания
 Фенилкетонурия — это наследственное заболевание, которое характеризуется нарушением белкового обмена.
Фенилкетонурия — это наследственное заболевание, которое характеризуется нарушением белкового обмена.
Впервые это заболевание обнаружили в 1934 году. Фенилкетонурия наследуется по так называемому аутосомно-рецессивному типу, то есть у совершенно здоровых родителей (носителей) могут родиться больные фенилкетонурией дети.
Виды фенилкетонурии
Существует 3 типа этого заболевания.
Фенилкетонурия первого типа – характеризуется дефицитом в организме фермента фенилаланин-4-гидроксилаз. Благодаря этому ферменту аминокислота фенилаланин превращается в тирозин. Чаще всего наследуется фенилкетонурия именно этого типа (в 98% случаев).
Фенилкетонурия второго типа – характеризуется недостатком такого фермента как дигидроптеридинредуктаза. Больные фенилкетонурией второго типа страдают судорогами и умственной отсталостью. Этот тип фенилкетонурии у детей встречается довольно редко (1-2%), но обычно приводит к смертельному исходу в 2-3-летнем возрасте.
Фенилкетонурия третьего типа – характеризуется дефицитом тетрагидробиоптерина. Симптомы фенилкетонурии этого типа включают в себя умственную отсталость вследствие микроцефалии – уменьшения объема мозга.
Причины фенилкетонурии
Основными причинами развития фенилкетонурии у детей считаются мутации гена, находящегося на 12 хромосоме. Родственные браки повышают риск возникновения аномалии.
При дефиците определенных ферментов наблюдается увеличение в крови производных фенилаланина, которые оказывают отравляющее действие на нервную систему ребенка. Фенилкетонурия наследуется примерно одинаково как мальчиками, так и девочками.
Симптомы фенилкетонурии
Симптомы фенилкетонурии проявляются не сразу. В большинстве случаев, заподозрить фенилкетонурию у детей сразу после рождения практически невозможно. На вид ребенок выглядит здоровым, рождается в срок и с нормальным весом. Но уже через несколько недель проявляются симптомы фенилкетонурии. Основным признаком заболевания считается сильная рвота. В период с двух до шести месяцев, как мама, так и лечащий доктор могут заметить отставание ребенка в психическом и физическом развитии.
Дети, больные фенилкетонурией, позже остальных сверстников начинают сидеть и ходить. Также явным симптомом фенилкетонурии является повышенная потливость с характерным «мышиным» запахом пота. Наблюдаются судороги, раздражительность, вялость, капризность и плаксивость, уменьшение размера головы, высыпания на коже. У детей, больных фенилкетонурией, поздно прорезываются зубы.
С развитием фенилкетонурии повышается мышечный тонус, что характеризуется определенной позой у ребенка, которую еще называют «поза портного» (согнутые в суставах руки и ноги).
Лечение фенилкетонурии
 Единственным лечением фенилкетонурии является соблюдение диеты, которой необходимо придерживаться более десяти лет после постановки диагноза. Дети, больные фенилкетонурией, не могут усваивать большое количество фенилаланина, поэтому имеются определенные нормы его потребления, которые зависят от возраста. Ребенок до двух месяцев может потреблять не более 60 мг/кг массы фенилаланина, а дети старше шести лет – не более 10-15 мг/кг.
Единственным лечением фенилкетонурии является соблюдение диеты, которой необходимо придерживаться более десяти лет после постановки диагноза. Дети, больные фенилкетонурией, не могут усваивать большое количество фенилаланина, поэтому имеются определенные нормы его потребления, которые зависят от возраста. Ребенок до двух месяцев может потреблять не более 60 мг/кг массы фенилаланина, а дети старше шести лет – не более 10-15 мг/кг.
Что касается грудного вскармливания, то тут необходимо придерживаться строгих правил. Мама должна контролировать количество грудного молока, которое выпивает ребенок. Поэтому для кормления необходимо использовать только сцеженное молоко и в количестве, которое разрешено для определенного возраста ребенка. Для этого существуют специальные таблицы с нормами потребления фенилаланина, а также формулы расчета количества грудного молока в день. Докармливают ребенка специальными смесями, которые не содержат фенилаланина.
Введение прикорма у детей с фенилкетонурией начинают с фруктовых и ягодных соков. В качестве твердой пищи предлагают ребенку овощные пюре без добавления молока. Используют безбелковые крупы и безмолочные каши на основе рисовой или кукурузной муки.
Лечение фенилкетонурии посредством питания включает в себя отказ или очень ограниченное потребление таких продуктов как рыба, мясо, хлебобулочные изделия, колбасы, творог, яйца, шоколад, бобовые, орехи и крупы. Эти продукты содержат большое количество белка. Фрукты и овощи вводят в рацион с учетом подсчета количества в них фенилаланина.
Лекарственное лечение фенилкетонурии предполагает прием витаминов, препаратов кальция, железа и фосфора. Также показан прием препаратов для улучшения мозгового кровообращения, микроциркуляции и тканевого обмена. Рекомендуют занятия лечебной физкультурой и массаж.
Видео с YouTube по теме статьи:
www.neboleem.net
Фенилкетонурия: лечение, причины, симптомы, признаки
- Беременность
- Развитие плода по неделям
- 1 триместр
- Скрининг 1 триместр
- 1-6 недели
- 1 неделя
- 2 неделя
- 3 неделя
- 4 неделя
- 5 неделя
- 6 неделя
- 7-12 недели
- 7 неделя
- 8 неделя
- 9 неделя
- 10 неделя
- 11 неделя
- 12 неделя
- 2 триместр
- Скрининг 2 триместра
- 13-18 недели
- 13 неделя
- 14 неделя
- 15 неделя
- 16 неделя
- 17 неделя
- 18 неделя
- 19-24 недели
- 19 неделя
- 20 неделя
- 21 неделя
- 22 неделя
- 23 неделя
- 24 неделя
- 3 триместр
- Скрининг 3 триместра
- 25-30 недели
- 25 неделя
- 26 неделя
- 27 неделя
- 28 неделя
- 29 неделя
- 30 неделя
- 31-36 недели
- 31 неделя
- 32 неделя
- 33 неделя
- 34 неделя
- 35 неделя
- 36 неделя
- 37-39 недели
- 37 неделя
- 38 неделя
- 39 неделя
- 37, 38, 39 недели у повторнородящих
- 1 триместр
- Как определить беременность
- Месячные и беременность
- Вопросы и рекомендации по беременности
- Выделения при беременности
- Питание при беременности
- Осложнения и боли при беременности
- Прерывание беременности
- Развитие плода по неделям
- Болезни
- Грипп Мичиган
- Рахит у грудничков
- Кишечная колика
- Пупочная грыжа
- Инструкции
- для детей
- при ОРВИ
- Виферон свечи
- Ибуклин Юниор
- Синупрет капли
- при кашле
- Аскорил сироп
- Бромгексин таблетки
- Пантогам сироп
- Синекод
- Саб симплекс
- Эриспирус сироп
- Эреспал сироп
- при гриппе
- Амоксиклав
- Амиксин
- Арбидол
- Панавир
- Ремантадин
- Тамифлю
- Циклоферон
- жаропонижающие
- Нурофен детский
- Панадол сироп
- Парацетамол сироп
- Цефекон свечи
- при болях
- Плантекс (от коликов)
- Смекта (от диареи)
- Энтерол (для кишечника) для детей
- Эспумизан беби (боли животика)
- Другие заболевания
- Вибуркол свечи (симптоматическое средство)
- Зиннат суспензия (отиты и т.п.)
- Изофра (риниты и синуситы)
- Мирамистин (антисептик)
- Отипакс (отит)
- Сиалор (ЛОР-заболевания)
- при ОРВИ
- при беременности и лактации
- при ОРВИ
- Анальгин
- Пиносол
- Тизин
- при кашле
- Биопарокс
- Гексорал спрей
- Лизобакт
- Либексин
- Сироп Алтея
- Стодаль
- Фарингосепт
- Цикловита
- при гриппе
- Арбидол
- Ацикловир
- Ремантадин
- Тамифлю
- жаропонижающие
- Ибупрофен
- при болях
- Ибупрофен
- Но-шпа
- Пенталгин
- Цитрамон
- Другие заболевания
- Клотримазол (грибок, инфекция половых органов)
- Линдинет 20 (противозачаточное)
- Мастодинон (нарушение менструации)
- Норколут (гормональный препарат)
- Полижинакс (противогрибковое)
- Тироксин (при гипотиреозе)
- Эстровэл (от женских заболеваний)
- Ярина (противозачаточное)
- при ОРВИ
- для взрослых
- при ОРВИ
- Амиксин
- Арбидол
- Гриппферон
- Интерферон
- Кипферон свечи
- Ротокан
- Ремантадин
- Синупрет
- Тамифлю
- при кашле
- АЦЦ
- Либексин
- Ренгалин
- Стоптуссин
- Стодаль
- при гриппе
- Амиксин
- Дибазол
- Кагоцел
- Лавомакс
- Ремантадин
- Флемоксин Солютаб
- Цефтриаксон
- жаропонижающие
- Индометацин
- Ибупрофен
- Ринза
- при болях
- Колофорт (для пищеварительного тракта)
- Кеторол
- Мовалис (противовоспалительное)
- Найз таблетки
- Тримедат (для кишечника)
- Невролгии, ЦНС, головной мозг
- Аспаркам
- Актовегин
- Комбилипен
- Нейромультивит
- Циннаризин
- Другие заболевания
- Адвантан (дерматологические заболевания)
- Азитромицин (ЛОР-заболевания)
- Диазолин (от аллергии)
- Лоратадин (при аллергии)
- Овесол (для печени)
- Эссенциале форте Н (для печени)
- Полидекса (ЛОР-заболевания)
- Клотримазол (грибок, инфекция половых органов)
- Циклоферон (инфекционные, бактериальные, грибковые заболевания)
- при ОРВИ
- для детей
- Грудное вскармливание
- Питание при ГВ
- Кормление грудью
- Таблетки при ГВ
- Болезни при ГВ
- Прикорм грудничка
- Вопросы и рекомендации
- Калькуляторы
- Калькулятор ХГЧ
- Спермограмма: расшифровка результата
- Календарь овуляции для зачатия
- Срок беременности по неделям и дням
- Дата родов по месячным, дате зачатия
- Календарь беременности по неделям
- Рассчитать пол ребенка
- Калькулятор роста и веса ребенка
Поиск
- Инструкции по применению (по алфавиту):
- А
- Б
- В
- Г
- Д
- Ж
- З
- И
- Й
- К
- Л
- М
- Н
- О
- П
- Р
- С
- Т
- У
- Ф
- Х
- Ц
- Э
- 0-9

- Беременность
- Развитие плода по неделям
- 1 триместр
- Скрининг 1 триместр
- 1-6 недели
- 1 неделя
- 2 неделя
- 3 неделя
- 4 неделя
- 5 неделя
- 6 неделя
- 7-12 недели
- 7 неделя
- 8 неделя
- 9 неделя
- 10 неделя
- 11 неделя
- 12 неделя
- 2 триместр
- Скрининг 2 триместра
- 13-18 недели
- 13 неделя
- 14 неделя
- 15 неделя
- 16 неделя
- 17 неделя
- 18 неделя
- 19-24 недели
- 19 неделя
- 20 неделя
- 21 неделя
- 22 неделя
- 23 неделя
- 24 неделя
- 3 триместр
- Скрининг 3 триместра
- 25-30 недели
- 25 неделя
- 26 неделя
- 27 неделя
- 28 неделя
- 29 неделя
- 30 неделя
- 31-36 недели
- 31 неделя
- 32 неделя
- 33 неделя
- 34 неделя
- 35 неделя
- 36 неделя
- 37-39 недели
- 37 неделя
- 38 неделя
- 39 неделя
- 37, 38, 39 недели у повторнородящих
- 1 триместр
- Как определить беременность
- Месячные и беременность
- Вопросы и рекомендации по беременности
- Выделения при беременности
- Питание при беременности
- Осложнения и боли при беременности
- Прерывание беременности
- Развитие плода по неделям
- Болезни
- Грипп Мичиган
- Рахит у грудничков
- Кишечная колика
- Пупочная грыжа
- Инструкции
- для детей
- при ОРВИ
- Виферон свечи
- Ибуклин Юниор
- Синупрет капли
- при кашле
- Аскорил сироп
- Бромгексин таблетки
- Пантогам сироп
- Синекод
- Саб симплекс
- Эриспирус сироп
- Эреспал сироп
- при гриппе
- Амоксиклав
- Амиксин
- Арбидол
- Панавир
- Ремантадин
- Тамифлю
- Циклоферон
- жаропонижающие
- Нурофен детский
- Панадол сироп
- Парацетамол сироп
- Цефекон свечи
- при болях
- Плантекс (от коликов)
- Смекта (от диареи)
- Энтерол (для кишечника) для детей
- Эспумизан беби (боли животика)
- Другие заболевания
- Вибуркол свечи (симптоматическое средство)
- Зиннат суспензия (отиты и т.п.)
- Изофра (риниты и синуситы)
- Мирамистин (антисептик)
- Отипакс (отит)
- Сиалор (ЛОР-заболевания)
- при ОРВИ
- при беременности и лактации
- при ОРВИ
- Анальгин
- Пиносол
- Тизин
- при ОРВИ
- для детей
zdorrov.com
Фенилкетонурия и нарушения обмена тетрагидробиоптерина у детей
Жалобы и анамнез
При отсутствии лечения на первом году жизни, обычно в возрасте 2−6 месяцев, родителей беспокоят вялость ребенка, отсутствие интереса к окружающему, иногда повышенная раздражительность, беспокойство, срыгивания, нарушение мышечного тонуса (чаще мышечная гипотония), признаки атопического дерматита, задержка психомоторного развития, иногда судороги. При своевременно назначенном патогенетическом лечении жалобы имеют более легкий характер или отсутствуют (Приложение В).Физикальное обследование
При отсутствии лечения обращают на себя внимание следующие фенотипические особенности: гипопигментация кожи, волос, радужной оболочки глаз, своеобразный «мышиный» запах мочи больных, возможно формирование микроцефалии. В психоневрологическом статусе отмечаются задержка статико-моторного и психоречевого развития, симптоматическая эпилепсия, а в некоторых случаях ― гидроцефалия.
Эпилептические приступы встречаются почти у половины нелеченых больных и в некоторых случаях могут служить первым признаком болезни. Обычно отмечаются генерализованные пароксизмы по типу инфантильных спазмов в виде «салаамовых судорог», кивков; могут наблюдаться абсансы. Приступы носят упорный характер и плохо поддаются антиконвульсантной терапии. При отсутствии патогенетического лечения болезнь медленно прогрессирует. Умственная отсталость, как правило, достигает глубокой степени: коэффициент умственного развития (intelligence quotient, IQ) составляет около 20 единиц (норма 85−115 единиц). В психологическом статусе больных отмечают нарушение игровой и предметной деятельности, отсутствие дифференцировки эмоциональных реакций, недостаточность экспрессивной и импрессивной речи. Могут наблюдаться двигательные, психопатоподобные и шизофреноподобные расстройства.
Аналогичные клинические симптомы, которые манифестируют после 2 месяцев жизни и достигают максимального проявления к 6 месяцам жизни, имеют Bh5-дефицитная ГФА6-PTPS, Bh5-дефицитная ГФАGTPSH, Bh5-дефицитная ГФА (тип С) вследствие недостаточности DHPR, Bh5-дефицитная ГФА вследствие недостаточности SR. Характерны прогрессирующее нарушение психомоторного развития, экстрапирамидные расстройства в виде изменения мышечного тонуса (гипотония мышц туловища и гипертонус мышц конечностей), тремор, атаксия, позднее ― нарушения походки, патологические рефлексы, гиперсаливация, нарушение терморегуляции, псевдобульбарные расстройства в виде затруднения глотания, поперхиваний во время приема пищи, микроцефалия, судороги, окулогирные кризы (эпизодическое содружественное отклонение глаз, обычно направленное вверх и латерально, редко вниз или строго латерально), экзема, гипопигментация кожи. У таких детей при рождении нередко отмечается низкая масса тела.
При недостаточности 6-PTPS различают два фенотипа. Наиболее часто (80%) встречающаяся тяжелая центральная (типичная) форма сопровождается резким дефицитом трансмиттеров и более выраженной тяжестью течения. Вторая, умеренная, периферийная форма сопровождается нормальным содержанием медиаторов, умеренной ГФА и умеренно выраженной клинической симптоматикой.
Для Bh5-дефицитной ГФА (тип D) вследствие недостаточности PCВD также характерны специфические изменения мышечного тонуса: постуральная нестабильность, гипокинезия, мышечная дистония (гипертонус конечностей, сниженный тонус мышц туловища).Лабораторная диагностика
• Рекомендуется проведение неонатального скрининга (определение концентрации фенилаланина в сухих пятнах крови) для доклинической диагностики ГФА и своевременного начала патогенетической терапии [1, 4−7] (Сила рекомендации A; уровень убедительности доказательств II ― здесь и далее Силу рекомендаций и уровни убедительности см. Критерии оценки качества медицинской помощи и Приложение А2).
Комментарии. Все формы ГФА можно диагностировать уже в первые недели или даже дни жизни ребенка, когда клинические проявления еще отсутствуют. Для этого проводят биохимический скрининг новорожденных на наличие ГФА. Подробная схема проведения неонатального скрининга представлена в Приложении Г2, описание правил взятия крови представлено в Приложении Г3.
• Рекомендуется использовать для неонатального скрининга методы флюориметрии или тандемной масс-спектрометрии [1, 4, 6, 7] (Сила рекомендации A; уровень убедительности доказательств II).
Комментарии. Флюориметрия ― количественный биохимический метод определения фенилаланина в крови с помощью современных автоматических флюориметров ― широко используется для проведения массового автоматизированного скрининга. Тандемная масс-спектрометрия ― аналитический метод исследования, основанный на определении отношения массы к заряду ионов, образующихся при ионизации исследуемых компонентов пробы, ― осуществляет качественную и количественную идентификацию аминокислот, позволяет одновременно определять уровни фенилаланина и тирозина, соотношение концентраций позволяет косвенно оценить степень снижения активности ФАГ.
• Главным критерием диагностики ГФА рекомендовано считать повышенное содержание фенилаланина в крови (Сила рекомендации A; уровень убедительности доказательств II).
Комментарии. Для всех указанных методов уровень ФА в крови человека выше 2,0 мг/дл (120 мкмоль/л) квалифицируют как ГФА. ГФА с уровнем выше 10,0 мг/дл (600 мкмоль/л) относят к различным формам ФКУ [6].
• Рекомендуется проведение дифференциальной диагностики ФКУ и других ГФА (второй этап скрининга) [1, 4, 5, 7, 8] (Сила рекомендации A; уровень убедительности доказательств II).
Комментарии. Второй этап скрининга необходим для выявления ВН4-зависимых форм ГФА и своевременного назначения патогенетической терапии. В настоящий момент для этого используется определение соотношения фенилаланин/тирозин (косвенно отражает наличие дефицита фермента РАН) и ДНК-диагностика [1, 7, 9, 10].
• При отсутствии данных неонатального скрининга диагностику заболевания рекомендовано осуществлять на основании совокупности генеалогического анамнеза, результатов клинического и биохимического обследования [1, 6, 8−11] (Сила рекомендации С; уровень убедительности доказательств II).
Комментарии. В этих случаях главным для установления диагноза остается биохимический критерий ― высокое содержание фенилаланина в сыворотке крови при отсутствии патогенетического лечения. В дальнейшем показано проведение молекулярной диагностики.
• Рекомендуется ДНК-диагностика с целью выявления мутаций в генах РАН и РТРS (Сила рекомендации A; уровень убедительности доказательств II).
Комментарии. ДНК-диагностика проводится с целью дифференциальной диагностики и для определения характера мутаций, что в дальнейшем помогает определить контингент пациентов для проведения теста на чувствительность к лечению сапроптерином. Существующие наборы позволяют определять частые мутации в гене РАН, имеющиеся у 80% больных ФКУ. При отсутствии исследуемых мутаций пациенту рекомендуется проведение секвенирования гена РАН. Также проводится ДНК-диагностика мутаций и секвенирование генов (PTPS, DHPR, PCD и др.), кодирующих известные ферменты различных стадий метаболизма Bh5.
В настоящее время известно более 900 мутаций в гене РАН, спектр и распространенность которых имеют этнические особенности. Для европеоидной расы мажорной мутацией в гене РАН является мутация R408W, в то время как в Японии и Китае данная мутация не найдена. Во многих европейских популяциях с относительно высокой частотой регистрируются следующие мутации: IVS12nt1, R261Q, R252W, R158Q, P281L, IVS10nt546, I65T.
Большинство генетических изменений гена РАН ― это миссенс-мутации во всех 13 экзонах гена или нетранслируемых фланкирующих участках гена, составляющие 59% всех вариантов. Также обнаружены мутации сплайсинга, нонсенс-мутации, мутации сдвига рамки считывания, более крупные делеции и инсерции. Разные мутации влияют на работу фермента РАН в различной степени: этим может объясняться большое разнообразие показателя фенилаланина в крови больных ФКУ. Большое количество мутаций гена РАН (например, R408W) приводят к резидуальной активности фермента ФАГ. При других мутациях (E390G, Y414C,A300S) толерантность к фенилаланину выше, и клиническая картина ФКУ менее выражена. Таким образом, по современным данным [12], результаты генотипирования при ФКУ потенциально обладают предиктивным значением.
Следует отметить, что при определенных мутациях в гене РАН, обусловливающих остаточную активность фермента ФАГ, введение кофактора Bh5 или его синтетических аналогов в терапию приводит к повышению или восстановлению активности ФАГ, что позволяет расширить диету с увеличением в рационе квоты натурального белка.
Инструментальная диагностика
• Рекомендовано проведение электроэнцефалографии для выявления паттернов гипсаритмии (даже при отсутствии клинических судорожных приступов), единичных и множественных спайк- и полиспайкразрядов, других изменений [1, 13] (Сила рекомендации С; уровень убедительности доказательств II).
• Рекомендовано проведение магнитно-резонансной томографии с целью выявления очагов перивентрикулярной лейкопатии, кортикальной атрофии и других изменений у пациентов старше 12 лет [1, 14, 15] (Сила рекомендации С; уровень убедительности доказательств II).
• Рекомендовано проведение ультразвукового исследования брюшной полости и почек для диагностики дискинезии желчных путей, диффузных изменений печени и поджелудочной железы, мочекаменной болезни [1, 7, 16] (Сила рекомендации С; уровень убедительности доказательств II).
• Рекомендовано проведение эзофагогастрофиброскопии для диагностики поражения слизистой оболочки желудка (по показаниям) [1, 7, 15, 16] (Сила рекомендации С; уровень убедительности доказательств II).
Иная диагностика (консультативная помощь)
• Рекомендовано психолого-педагогическое консультирование и логопедическое тестирование [1, 7, 13, 17] (Сила рекомендации С; уровень убедительности доказательств II).
Комментарии. Тестирование проводится с целью определения уровня интеллектуального и речевого развития, возможностей социальной адаптации и составления индивидуального плана психолого-педагогического сопровождения.
Рекомендовано проведение по показаниям консультаций специалистов (невропатолога, гастроэнтеролога, офтальмолога, аллерголога и др.) [7, 16, 18] (Сила рекомендации С; уровень убедительности доказательств II).
diseases.medelement.com
что это такое? Симптомы, диагностика и лечение. Как наследуется фенилкетонурия :: SYL.ru
Сейчас диагностируется огромное количество наследственных заболеваний, которые ребенок получает от отца или от матери. Экологическая обстановка, неправильное питание, нездоровый образ жизни – все это приводит к тому, что клетки мутируют, генетическая информация претерпевает существенные изменения. Вот от этого и возникает огромное количество наследственных заболеваний. Одним из них является фенилкетонурия. Что это за болезнь, знают не многие, поэтому постараемся во всем разобраться.
Сущность понятия
Фенилкетонурия является наследственным заболеванием, связано оно с серьезными нарушениями в белковом обмене. Это, в свою очередь, приводит к поражению нервной системы.
Неработоспособность всего одного фермента, фенилаланина, а в результате – такие серьезные проблемы со здоровьем, как фенилкетонурия. Что это за состояние, когда в организме происходит накопление большого количества токсических веществ? Все ядовитые соединения запасаются в биологических жидкостях, поэтому для медиков диагностировать болезнь обычно не составляет особого труда.
Если вовремя не принять меры, то можно наблюдать серьезные поражения нервной системы, а это уже приводит к нарушениям в функционировании всего организма.
Таким образом, без соответствующего лечения о нормальной жизни пациента не может быть и речи.
Причины заболевания
Все белки состоят из аминокислот, которых всего 20, но среди них имеются такие, которые синтезируются в организме человека. Некоторые же должны поступать только извне. К незаменимым аминокислотам относится и фенилаланин. У здорового человека он, попадая внутрь, превращается в тирозин. Это совершенно другая аминокислота, и только несколько процентов вещества направляется в почки и там преобразуется в фенилкетон, достаточно токсичное вещество.
Если у человека отсутствует фермент фенилаланин-4-гидроксилаза или неправильно работает тот, который превращает фенилаланин в иное вещество, то вот в этом случае и развивается фенилкетонурия. Что это достаточно серьезный симптом, скажет вам каждый врач, поэтому необходимо принимать срочные меры.
А привести к отсутствию нужного фермента может мутация гена в хромосоме 12.
Разновидности фенилкетонурии
Если рассматривать формы заболевания, то они могут быть такими:
- Классическая. В этом случае мы наблюдаем, что фенилкетонурия – рецессивный признак. Встречается такая форма у одного ребенка на десять тысяч здоровых детей. Если не принять меры, то едва ли больной человек доживет до тридцати лет.
- Вариативная форма. Не передается по наследству, а вызывает ее мутация в генах. Течение ее более тяжелое, а ранняя смертность прогнозируется с вероятностью практически 100%.

Кроме форм, врачи различают еще и типы фенилкетонурии:
- Первый тип характеризуется тем, что наблюдается недостаток фермента фенилаланин-4-гидроксилазы, который и отвечает за превращение фенилаланина. В 98% случаев диагностируется именно он.
- Второй. Его отличает малое содержание фермента дигидроптеридинредуктазы. У таких больных наблюдаются судороги, а также умственная отсталость. Несмотря на свою редкую встречаемость, смертность от этого типа может наступать в 2-3-летнем возрасте.
- Третий тип характеризуется тем, что возникает дефицит тетрагидробиоптерина. В результате происходит уменьшение объемов мозга, что приводит к умственной отсталости.
Симптоматика болезни
Сразу после рождения ребенка по его внешнему виду или поведению трудно диагностировать заболевание. Основные признаки начнут проявляться немного погодя. Однако еще в родильном доме медики вполне способны поставить диагноз «фенилкетонурия». Симптомы у этого заболевания следующие:
- частая рвота без видимых причин;
- плаксивость;
- вялость;
- могут появляться высыпания по всему телу;
- моча имеет «мышиный» запах;
- ребенок отстает в физическом и умственном развитии от своих сверстников.
Достаточно взять анализ крови и мочи, чтобы поставить правильный диагноз.
Признаки фенилкетонурии
Постепенно при отсутствии надлежащего лечения у больного можно будет наблюдать такие признаки:
- Судорожный синдром. Он начинает проявляться в раннем детстве и сохраняется у взрослых.
- Недостаток пигмента в коже и волосах. Поэтому такие пациенты, как правило, светловолосые и имеют белую кожу.
- Воспалительные процессы, которые по незнанию можно принять за аллергическую реакцию.

Первые признаки умственного отставания можно заметить у ребенка уже в возрасте полугода. Он перестает запоминать новую информацию, и кажется, что он совершенно не способен к обучению. Насторожиться родители должны и тогда, когда малыш забывает то, чему уже давно научился, например, как держать ложку, сидеть, играть с погремушкой. Тревогу нужно бить и в том случае, если ребенок перестает узнавать родителей и близких людей, а также чрезмерная плаксивость с возрастом не проходит.
Вот признаки, которые имеет фенилкетонурия, симптомы болезни необходимо рассматривать только в комплексе, потому что по отдельности они вполне могут встречаться и у здоровых детей.
Выявление заболевания
Поставить правильный диагноз можно двумя путями:
- Провести анализ крови и мочи у новорожденного еще в родильном доме. это, как правило, делают во всех случаях.
- Определить наличие фенилкетонов в биологических жидкостях взрослого человека при наличии соответствующих признаков.
У детей в роддоме кровь берут на 4-5-й день и определяют содержание фенилаланина. Если таковой обнаруживается, то ребенка с мамой направляют на консультацию к генетику.
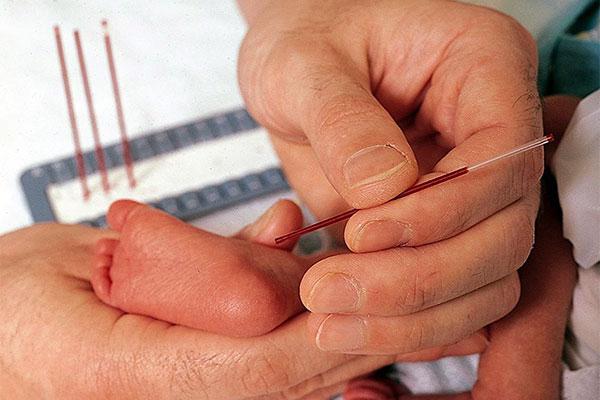
Перед выпиской обязательно поинтересуйтесь, делали ли вашему ребенку анализ на фенилкетонурию. Несмотря на небольшую встречаемость этого заболевания, лучшим решением будет все равно подстраховаться.
Наследование
Так как фенилкетонурия наследуется как рецессивный признак, то для проявления его у ребенка необходимо, чтобы у обоих родителей имелся дефектный ген. Именно поэтому родственные браки во многих странах запрещены.
Если рассматривать случай рождения детей в обычной семье, то у носителей такой мутации они могут быть:
- 25% вероятности, что ребенок родится больным.
- В 50% случаев малыш здоров, но является носителем дефектного гена.
- Четвертая часть потомства будет абсолютно нормальной.
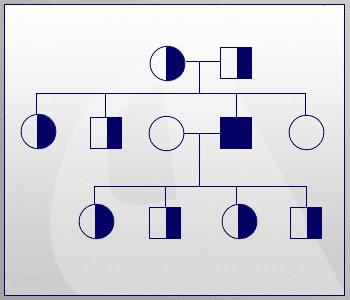
Эта схема не дает полной картины рождаемости больных детей. Она только отражает вероятность, поэтому у каждой семейной пары процент дефектных генов может быть свой, и предсказать исход, к сожалению, невозможно. Сейчас существуют консультации, в которых специалисты-генетики помогают парам спрогнозировать рождение у них больного ребенка, рассказывая при этом, как фенилкетонурия наследуется.
Лечение
Как только ребенку поставлен такой диагноз, меры необходимо принимать незамедлительно. В первую очередь, нужно исключить из рациона белковые продукты. Соблюдать такое строгое ограничение необходимо до 10-12 лет, а лучше и всю жизнь.
Так как младенцы находятся на грудном вскармливании и обычно ничего, кроме материнского молока, еще не употребляют, то врачи рекомендуют маме сократить его потребление ребенком. Сделать это возможно только при одном условии: давать малышу сцеженное молоко, чтобы точно видеть его количество.
Докармливать придется смесями, которые в своем составе не имеют фенилаланина. Когда наступит время введения прикорма, необходимо выбирать пюре и каши без добавления молока. Можно давать соки, овощные пюре.
Врач обязательно назначает и медикаментозное лечение. Обычно это препараты, которые содержат фосфор, ведь этот элемент не зря считают «элементом жизни и мысли», так как он играет важную роль в работе нашего мозга. Также назначаются лекарства, содержащие железо, кальций, они помогают улучшить кровообращение и мозговую деятельность.
Лечение не должно сводиться к полному исключению фенилаланина, так как в этом случае может наблюдаться его недостаток, что приводит к упадку сил, потере аппетита. Кроме того, начинается понос и возникают высыпания на коже.
Чтобы узнать, насколько лечение эффективно, следует периодически сдавать анализ крови и мочи на содержание фенилаланина.
Заболевание у детей
Именно в детском возрасте организм развивается такими темпами, которых не будет в другие периоды жизни. Поэтому в это время важно принять все меры для нормального развития нервной системы. Дети, больные фенилкетонурией, нуждаются не только в приеме медицинских препаратов и специальном питании, но и особом к себе отношении.
Прежде всего, это постоянное внимание, чтобы от зоркого глаза матери не укрылись малейшие отклонения в развитии. Можно применять следующие методы лечения:
- лечебная физкультура, которая поможет ребенку нормально развиваться физически;
- массаж;
- психологическая помощь;
- коррекционная педагогика.

Родители должны понимать, что жизнь и здоровье их ребенка будет в большей степени зависеть от них самих. Какую обстановку они смогут создать вокруг больного малыша, насколько точно будут соблюдены рекомендации врачей по питанию, будут ли близкие люди реагировать на отклонения в умственном и физическом развитии – все эти моменты очень важны.
Народная медицина для избавления от недуга
Народные рецепты находят свое применение при лечении многих болезней. Не исключение и фенилкетонурия. Что это заболевание потребует пересмотра всего образа жизни – это факт. Ребенок должен расти и иметь представление о своем недуге. Родители обязаны в доступной форме объяснить ему, когда он будет способен осознавать полученную информацию, насколько серьезно его положение. Диету и лечение необходимо соблюдать в течение всей жизни. Только в этом случае можно гарантировать полноценное существование.
Народные лекари при фенилкетонурии рекомендуют употреблять больше растительных белков. В такой пище фенилаланина гораздо меньше, чем в животных продуктах. Не запрещается в рацион вводить фрукты и овощи. В них много витаминов и микроэлементов, которые в обязательном порядке необходимы для нормальной работы нервной системы. То есть народная медицина придерживается мнения, что такому больному желательно соблюдать вегетарианскую диету.
Питание при фенилкетонурии
Фенилаланин содержится практически во всех продуктах, в которых есть белок. Необходимо постараться исключить их из своего рациона, и в первую очередь это касается молока и мяса.
Если поставлен диагноз «фенилкетонурия», питание должно быть пересмотрено в первую очередь. Все продукты при этом можно разделить на несколько групп:
- Разрешены к использованию всегда: картофель, сахар, чай, растительные масла.
- Можно употреблять в небольшом количестве: рис, мед, масло сливочное, хлебобулочные изделия, овощи и фрукты.
Полностью необходимо исключить из своего меню: яйца, рыбу и мясо, молоко, макароны, бобовые, орехи, кукурузу, молочные продукты, шоколад.

Учитывая тот факт, что фенилаланин превращается в здоровом организме в тирозин, то больные фенилкетонурией должны в рацион включать продукты, содержащие его в достаточном количестве. К такой пище можно отнести грибы и растительные составляющие.
Прогноз на будущее при фенилкетонурии
Очевидно, что данное заболевание требует немедленного принятия мер, в противном случае жизнь человека будет короткой.
Заболевание «фенилкетонурия» требует внимательного отношения к пациенту. Если соблюдать строгую диету и выполнять все рекомендации врачей, то ребенок сможет нормально расти и развиваться. Прогноз будет также зависеть от того, какие заболевания сопутствуют генетическому недугу и есть ли другие патологии.
Постепенно, с возрастом, организм может в какой-то мере приспосабливаться к повышенному содержанию фенилаланина, поэтому можно иногда допускать послабления в диете. Главное, чтобы не увлечься этими слабостями и вовремя остановиться и перейти на правильное питание.
Если женщина страдает этим недугом, то ей придется еще строже относиться к соблюдению всех рекомендаций, ведь она – будущая мама. Только в этом случае у нее есть возможность родить здорового ребенка.
Это особенно актуально, если учитывать, что методов профилактики данного заболевания практически не существует.
www.syl.ru
— фенилкетонурии — Биохимия
При любых нарушениях превращения фенилаланина в тирозин развивается фенилкетонурия.
По Mc Kusick выделяется несколько типов фенилкетонурии: классическая (1 типа), вариантная (2 типа), 3 типа и материнская.
Фенилкетонурия 1 типа (классическая)
Фенилкетонурия 1 типа является наиболее распространенной аминоацидопатией. Частота ФКУ среди новорожденных по данным массового скрининга в различных странах составляет в среднем 1:10000, однако значительно варьирует в зависимости от популяции: от 1:4560 в Ирландии, до 1:100000 в Японии.
Этиология
Заболевание наследуется аутосомно-рецессивно и вызвано мутацией, которая вызывает снижение активности фермента фенилаланин-4-монооксигеназы, обеспечивающей превращение фенилаланина в тирозин. Фермент имеется только в печени, почках, поджелудочной железе.
Патогенез
В патогенезе ФКУ имеют значение многие обстоятельства, в частности:
- значительное накопление в тканях и жидкостях больного организма фенилаланина и его производных (фенилпировиноградная, фенилмолочная (миндальная), фенилуксусная, гиппуровая кислоты, фенилэтиламин, фенилацетилглютамин) и вызванный ими ацидоз,
- прямое токсическое действие указанных веществ на центральную нервную систему, которое заключается в торможении фенилаланином активности ряда ферментов, в том числе пируваткиназы (окисление глюкозы), тирозиназы (синтез меланина), тирозин-гидроксилазы (синтез катехоламинов) и нарушение синтеза моноаминовых нейромедиаторов – тирамина, октопамина,
- нарушение синтеза серотонина, т.к. фенилаланин-4-монооксигеназа также вовлечена в гидроксилирование триптофана до 5-гидрокситриптофана, предшественника серотонина,
- конкурентное снижение фенилаланином транспорта в клетки ароматических аминокислот – триптофана и тирозина,
- нарушение синтеза простых и сложных белков в тканях, что вызывает тяжелые повреждения мозга и нарушение функции печени у большинства больных.
Превращение фенилаланина при фенилкетонурии
Клиническая картина
Ребенок с фенилкетонурией выглядит при рождении здоровым. Манифестация ФКУ происходит на первом году жизни, обычно в возрасте 2-6 мес. Первым симптомом заболевания может стать рвота. Другими ранними проявлениями болезни служат вялость ребенка, чрезмерная сонливость, отсутствие интереса к окружающему, иногда повышенная раздражительность, беспокойство, плаксивость, также отмечаются срыгивания, нарушение мышечного тонуса (чаще мышечная гипотония), судороги.
Характерным признаком является повышенная потливость, от мочи и пота исходит необычный запах фенилуксусной кислоты, который характеризуют как заплесневелый, мышиный или волчий.
Дети отстают в физическом и нервно-психическом развитии.
В связи с высокой частотой фенилкетонурии, тяжестью ее проявлений и возможностью профилактического лечения в России принято неонатальное обследование детей в роддомах в возрасте 4-5 дней.
У больных детей задержка психического и речевого развития может происходить постепенно и стать очевидной лишь через несколько месяцев. Нелеченный ребенок теряет около 50 баллов IQ к концу первого года жизни. Только 4-5% остаются на стадии дебильности, остальные имеют коэффициент интеллекта <60 и являются имбецилами или идиотами.
В старшем возрасте нелеченные дети становятся гиперактивными, осуществляют бесцельные движения, ритмические покачивания, у них определяется атетоз (мучительные непроизвольные движения кистей рук, языка, лица и др.). Дети имеют глубокую задержку моторного развития – 30% не ходит, 60% не говорит.
Основы лечения
Вовремя начатое лечение (диетотерапия) обеспечивает хороший клинический эффект, нормальную продолжительность жизни.
Единственным методом лечения является диетотерапия – исключение из питания больного высокобелковых продуктов питания с высоким количеством фенилаланина (мясо, рыба, яйцо, молоко, крупы). Вместо натурального белка используют специальные гидролизаты белка, частично или полностью лишенные фенилаланина.
Однако больной должен получать с пищей определенные количества фенилаланина, покрывающие минимальную суточную потребность, что составляет 50-60 мг/кг для детей первого года жизни и 15-40 мг/кг для детей более старшего возраста. Это количество фенилаланина поступает за счет овощей, фруктов и других продуктов. Нельзя полностью исключать из рациона новорожденного материнское молоко, так как оно обеспечивает иммунитет ребенка в первые 6 месяцев жизни.
Строгое ограничение белков животного происхождения требуется на протяжении первых 2-3 лет жизни или как минимум до 6 лет, 5-10 лет, или до периода полового созревания (по разным данным). Во время беременности больные женщины должны возвращаться к диете, чтобы не допустить развития у ребенка умственной отсталости.
Больные нуждаются в дополнительном введении витаминов, особенно группы В, минеральных веществ и микроэлементов.
Фенилкетонурия 2 типа
Этиология
Аутосомно-рецессивный дефект дигидробиоптеринредуктазы.
В результате недостаточности фермента нарушается восстановление активной формы тетрагидробиоптерина, участвующего в качестве кофактора гидроксилаз фенилаланина и триптофана. Вследствие этого нарушается превращение фенилаланина в тирозин, триптофана в 5-гидрокситриптофан.
Патогенез
Отмечается снижение уровня фолатов в сыворотке крови, эритроцитах и цереброспинальной жидкости. Это объясняется тесной взаимосвязью обмена фолатов и биоптерина, в частности участием дигидробиоптеринредуктазы в метаболизме тетрагидрофолиевой кислоты.
Клиническая картина
В клинической картине преобладает тяжелая умственная отсталость, судороги, признаки повышенной возбудимости, сухожильная гиперрефлексия, мышечная дистония и гипотония, хореиформные движения (непроизвольные трясущиеся движения головы, лица или конечностей), спастический тетрапарез.
Течение болезни прогрессирующее и нередко приводит к смерти в 2-З-летнем возрасте. Появление клинической симптоматики, как правило, развивается в начале второго полугодия жизни, не смотря на диетотерапию.
Основы лечения
В отличие от классической формы этот вариант не поддается лечению ранним ограничением содержания фенилаланина в пище
Лечение тетрагидробиоптерином неэффективно, так как он не проникает через гематоэнцефалический барьер. Заместительная терапия L-ДОФА и 5-гидроокситриптофаном частично обходит блок в синтезе дофамина и серотонина.
Фенилкетонурия 3 типа
Этиология
Заболевание наследуется аутосомно-рецессивно и связано с недостаточностью 6‑пирувоил-тетрагидроптеринсинтазы, участвующей в процессе синтеза тетрагидробиоптерина из дигидронеоптерин-трифосфата.
Патогенез
Ключевую роль в патогенезе играет нарушение синтеза тетрагидробиоптерина. Развивающиеся при этом расстройства сходны с нарушениями, наблюдаемыми при ФКУ II.
Клиническая картина
Неврологические нарушения, в частности мышечная гипотония и задержка двигательного развития, появляются раньше, чем при классической ФКУ. Даже при адекватном снижении уровня фенилаланина в крови с помощью диеты, нарастание клинической симптоматики не прекращается. В отличие от больных ФКУ-II у этих больных судорог не отмечается (причина не ясна).
Основы лечения
Лечение тетрагидробиоптерином неэффективно, так как он не проникает через гематоэнцефалический барьер. Заместительная терапия L‑ДОФА и 5‑гидроокситриптофаном частично обходит блок в синтезе дофамина и серотонина. Диета неэффективна.
Другие варианты ФКУ
Также известны другие формы атипичной ФКУ, связанные с дефицитом тетрагидробиоптерина.
Недостаточность гуанозинтрифосфат-циклогидролазы описана по крайней мере у пяти больных. Этот фермент катализирует первую ступень синтеза тетрагидробиоптерина из ГТФ и при его дефиците в моче обнаруживается крайне низкая концентрация всех птеринов.
Материнская ФКУ
Этиология
Появление умственной отсталости среди потомства женщин с ФКУ, не соблюдающих диету в зрелом возрасте, получило наименование материнской ФКУ.
Патогенез
Патогенез патологии мало изучен, однако предполагается, что он сходен с патогенезом остальных форм ФКУ. Тяжесть поражения плода коррелирует с уровнем фенилаланина в плазме крови матери. И в связи с накоплением этой аминокислоты в плаценте ее содержание в организме плода оказывается выше, чем у матери. Тем не менее, прямое токсическое действие фенилаланина точно не подтверждено.
biokhimija.ru