
18 хромосома синдром эдвардса фото: Трисомия 18: Синдром Эдвардса Статьи
Трисомия 18: Синдром Эдвардса Статьи
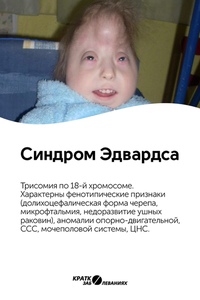
Синдром Эдвардса – это редкое генетическое заболевание, связанное с дублированием 18 хромосомы. Впервые описано это заболевание было в 1980 году доктором Джоном Эдвардсом, который описал симптомы. Трисомия 18 может выражаться в полной и в мозаичной форме. При этом мозаичная форма синдрома имеет более благоприятный прогноз.
Диагностика синдрома
В связи с появлением современных технологий, сейчас возможно раннее прогнозирование синдрома Эдвардса – начиная с ранних сроков беременности. В случае, если установлен синдром, решение о сохранении беременности принимает женщина. В редких случаях генетическая патология не выявляется до момента появления ребенка на свет. После рождения ребенка трисомия 18 может устанавливаться по внешним признакам, однако точный ответ может дать только кариотипический анализ. Он же поможет определить, полная форма синдром у больного, или мозаичная. В случае подозрения на синдром Эдвардса, пациент нуждается в консультации генетика с последующей сдачей анализов.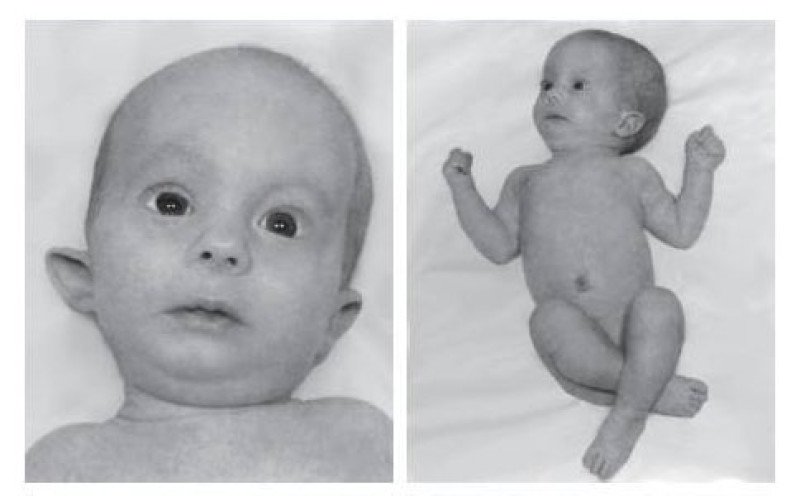
Признаки трисомии 18
Первые признаки генетического отклонения у плода появляются еще во время беременности. Вот самые частые из них: сильная задержка внутриутробного развития на несколько недель, пороки развития головного мозга, многоводие, слабые шевеления плода, наличие только одной пупочной артерии. Подобная беременность часто длится 42 недели и более, а ребенок при этом рождается с небольшим весом.
Характерные для синдрома Эдвардса внешние признаки сразу заметны даже у новорожденного ребенка. Такой ребенок будет иметь низкую массу тела, долихоцефалический череп, низкий лоб и широкий затылок, недоразвитые и низкорасположенные ушные раковины. Небо у таких детей обычно высокое, «готическое», верхняя губа короче, чем у обычных детей. Может встречаться множество различных изменений опорно-двигательного аппарата: расширенная грудная клетка, узкий таз, вывихи бедер, стопы-«качалки». Пальцы на ногах часто сросшиеся или имеют перепонки. Пальцы рук скрещены и укорочены.
Часто встречаются проблемы с недоразвитием или неправильным развитием органов пищеварения и мочеполовой системы.
Такие дети обычно имеют проблемы с глотанием, сосанием. Нередки случаи рефлюкса и очень часто требуется зондовое выкармливание подобных детей. Многие дети с трисомией 18 предрасположены к возникновению судорог, а также – к необъяснимым скачкам температуры без других симптомов.
Вес у таких детей прибавляется очень плохо, нередко к году ребенок с синдромом Эдвардса не доходит до планки даже в 5 кг.
Умственное развитие детей с синдромом Эдвардса обычно соответствует тяжелым степеням олигофрении, однако дети с мозаичной формой синдрома иногда добиваются определенных успехов в умственном развитии.
Прогноз для людей с синдромом Эдвардса
Как и любое другое генетическое заболевание, синдром Эдвардса не лечится. Однако возможно симптоматическое лечение, связанное с облегчением жизни данному ребенку, в том числе и хирургическое лечение пороков, сопутствующих синдрому.
Прогноз для жизни детей, у которых стоит диагноз «синдромом Эдвардса», в России в настоящее время неблагоприятен. Лишь небольшой процент детей с полной формой синдрома доживает до года. Средний срок жизни мальчиков ограничивается 2-3 месяцами, девочек – 10 месяцами. Однако при хорошем уходе со стороны родителей и внимательном наблюдении врачей дети с синдромом Эдвардса могут прожить гораздо дольше и иметь другое качество жизни. Они вполне могут научиться узнавать родных, улыбаться, самостоятельно кушать – при условии внимательного и заботливого к ним отношения. Дети с мозаичной формой синдрома имеют еще больше шансов на продолжительную и более полноценную жизнь.
В Европе и Америке люди с полной формой синдрома Эдвардса доживают до совершеннолетия. В России таких случаев пока зарегистрировано не было. Однако медицина не стоит на месте, и образованность людей повышается, поэтому вполне возможно, что скоро прогнозы для жизни людей с синдромом Эдвардса в России станут более благоприятными.
Синдром Эдвардса, ДНК-диагностика синдрома Эдвардса
Синдром Эдвардса (трисомия по хромосоме 18) — второе по частоте после синдрома Дауна хромосомное нарушение. Частота синдрома Эдвардса составляет 1:5000-1:7000 новорождённых. Девочки с синдромом Эдвардса рождаются в три раза чаще мальчиков.
| Рисунок 1. Пример диагностики синдрома Эдвардса методом КФ-ПЦР. | Рисунок 2. Больной ребенок с синдромом Эдвардса. |
Пример диагностики синдрома Эдвардса (трисомии по хромосоме 18). Фиолетовым цветом выделены маркеры, расположенные в районе хромосомы 18, критическом для развития синдрома Эдвардса. В генотипе исследуемого образца по маркеру D18S978 наблюдается 1 пик (маркер неинформативен), по маркерам D18S535 и D18S386 — 3 пика (трисомия), по маркерам D18S390 и D18S819 — эффект дозы – неравное соотношение высоты двух пиков (трисомия). |
|
«Золотым стандартом» выявления хромосомных нарушений во всем мире долгое время являлся и продолжает оставаться метод кариотипирования с дифференциальной окраской хромосом. Этот метод позволяет анализировать кариотип в целом и определять крупные (не менее 5-10 млн пар нуклеотидов) хромосомные перестройки. Однако у него существует ряд ограничений, таких как трудоемкость, длительность (1-2 недели), высокие требования к квалификации и опыту специалиста, проводящего исследование, а также, в ряде случаев, технические проблемы (недостаточное количество и качество исследуемого материала, отсутствие митозов или роста культуры).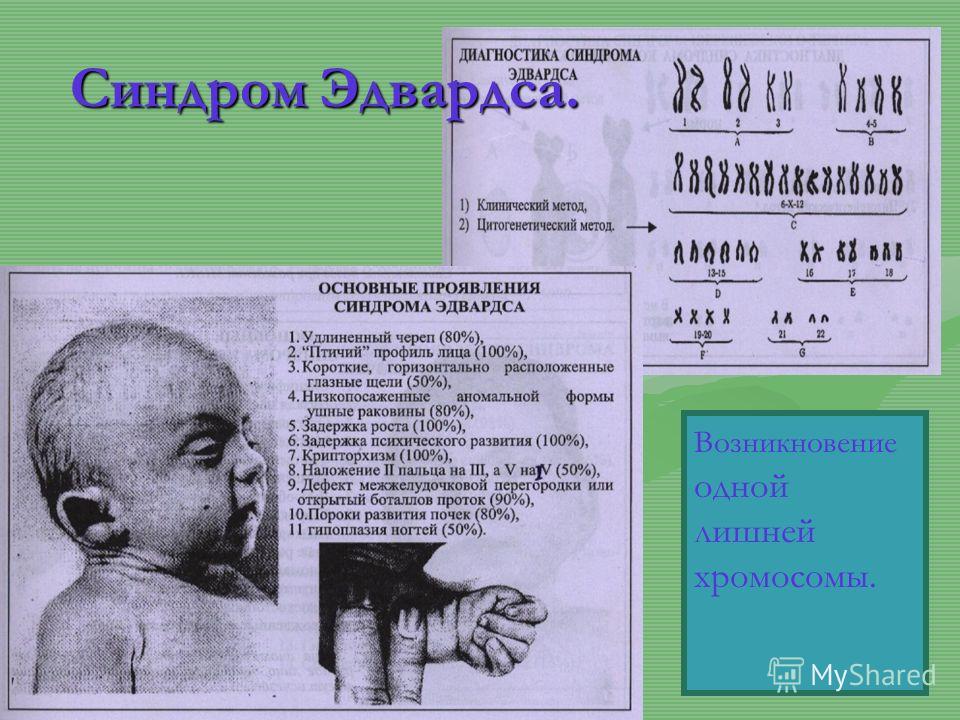
Этих недостатков лишен метод количественной флуоресцентной полимеразной цепной реакции (КФ-ПЦР), который все более широко применяется для диагностики анеуплоидий, в том числе и синдрома Эдвардса (Рис. 1). Этот метод обладает достоверностью, сравнимой с достоверностью стандартного кариотипирования, является более быстрым, дешевым, менее требовательным к количеству и качеству материала (поскольку не связан с ростом культуры клеток) и позволяет одновременно анализировать большое число образцов. Однако метод КФ-ПЦР имеет и ограничения: в мозаичных случаях он позволяет выявлять только высокоуровневый мозаицизм (от 20%), кроме того, он не может исключить наличие более редких хромосомных нарушений, которые могут быть связаны с пороками развития плода. При проведении дородовой диагностики синдрома Эдвардса, кроме материала плода, необходимо предоставлять биологический материал матери для того, чтобы исключить возможность получения ложноотрицательного результата из-за неправильного забора плодного материала.

Причиной развития синдрома Эдвардса является утроение хромосомы 18. Трисомия по хромосоме 18 является частным случаем анеуплоидии– наличия в геноме набора хромосом, отличного от стандартного для данного вида и некратного ему. Трисомия хромосомы 18 обычно вызвана нерасхождением хромосом при формировании половых клеток родителя (яйцеклеток и сперматозоидов), в результате чего ребенок получает от матери или от отца лишнюю 18-ю хромосому. В этом случае все клетки организма ребёнка будут нести аномалию. В том случае, когда нерасхождение хромосом возникает при делении какой-либо клетки зародыша, наблюдается мозаичный вариант синдрома Эдвардса (10% случаев).
Риск рождения детей с синдромом Эдвардса, по разным литературным данным, не изменяется или незначительно возрастает с увеличением возраста беременной женщины.
Пренатальная диагностика синдрома Эдвардса включает в себя два этапа. На первом этапе, на сроке беременности 11-13 недель, проводится скрининг, который основывается преимущественно на биохимических показателях, поскольку на ранних сроках УЗИ не позволяет обнаружить в случае синдрома Эдвардса каких-либо грубых аномалий развития, которые могут быть выявлены лишь к 20-24 неделе. Биохимический анализ уровня определенных белков в крови беременной женщины (свободной β-субъединицы хорионического гормона человека (β-ХГЧ) и ассоциированного с беременностью плазменного протеина А (pregnancy associated plasma protein-A, РАРР-А)), с учетом ее возраста, позволяет рассчитать для нее риск рождения больного ребенка. Однако эти методы не позволяют поставить точный диагноз, и в результате проведенного скрининга лишь формируется группа риска беременных с повышенной вероятностью рождения больного синдромом Эдвардса. На втором этапе в группе риска проводится инвазивная процедура для получения плодного материала, необходимого для точного определения статуса плода. В зависимости от срока беременности это может быть биопсия ворсин хориона (8-12 недели), амниоцентез (14-18 недели) или кордоцентез (после 20-й недели). В полученных образцах ткани плода проводится определение хромосомного набора.
Биохимический анализ уровня определенных белков в крови беременной женщины (свободной β-субъединицы хорионического гормона человека (β-ХГЧ) и ассоциированного с беременностью плазменного протеина А (pregnancy associated plasma protein-A, РАРР-А)), с учетом ее возраста, позволяет рассчитать для нее риск рождения больного ребенка. Однако эти методы не позволяют поставить точный диагноз, и в результате проведенного скрининга лишь формируется группа риска беременных с повышенной вероятностью рождения больного синдромом Эдвардса. На втором этапе в группе риска проводится инвазивная процедура для получения плодного материала, необходимого для точного определения статуса плода. В зависимости от срока беременности это может быть биопсия ворсин хориона (8-12 недели), амниоцентез (14-18 недели) или кордоцентез (после 20-й недели). В полученных образцах ткани плода проводится определение хромосомного набора.
В Центре Молекулярной Генетики проводится диагностика синдрома Эдвардса (в том числе и пренатальная) методом КФ-ПЦР.
СИНДРОМ ЭДВАРДСА КАК ПРОЯВЛЕНИЕ ГЕНЕТИЧЕСКОГО ЗАБОЛЕВАНИЯ В ПЕДИАТРИЧЕСКОЙ ПРАКТИКЕ (клинический случай) Текст научной статьи по специальности «Клиническая медицина»
JOURNAL OF NEW MEDICAL TECHNOLOGIES, eEdition — 2018 — N 2
УДК:61 DOI: 10.24411/2075-4094-2018-16008
СИНДРОМ ЭДВАРДСА КАК ПРОЯВЛЕНИЕ ГЕНЕТИЧЕСКОГО ЗАБОЛЕВАНИЯ В ПЕДИАТРИЧЕСКОЙ ПРАКТИКЕ
(клинический случай)
Е.С. ЗАХАРОВА, А.Д. ЛАРИКОВА
ФГБОУ ВПО «Тульский государственный университет», медицинский институт, ул. Болдина, д. 128, Тула, 300028, Россия
Аннотация. В данной статье освещена тема редких генетических заболеваний в практике врача-педиатра на примере синдрома Эдвардса. Также представлен разбор клинического случая выявления данного синдрома у ребенка, находившегося на стационарном лечении в отделении выхаживания недоношенных детей. Врачу-педиатру необходимо и очень важно помнить о наличии такого грозного заболевания для своевременной диагностики и выбора тактики дальнейшего ведения таких пациентов. В статье представлены данные об этиологии, эпидемиологии, клинических признаках, диагностике и дальнейшей тактики лечения данной группы пациентов.
Также представлен разбор клинического случая выявления данного синдрома у ребенка, находившегося на стационарном лечении в отделении выхаживания недоношенных детей. Врачу-педиатру необходимо и очень важно помнить о наличии такого грозного заболевания для своевременной диагностики и выбора тактики дальнейшего ведения таких пациентов. В статье представлены данные об этиологии, эпидемиологии, клинических признаках, диагностике и дальнейшей тактики лечения данной группы пациентов.
Синдром Эдвардса (другое название-синдром трисомии хромосомы 18) — генетическое заболевание, обусловленное наличием у человека дополнительной копии восемнадцатой хромосомы, то есть вместо двух 18-х хромосом в норме у больного присутствуют три 18-х хромосомы. Синдром Эдвардса является вторым по частоте хромосомным заболеванием после синдрома Дауна и характеризуется множеством пороков внутриутробного развития ребенка.
Описание основных симптомов данной болезни было сделано еще в начале XX века.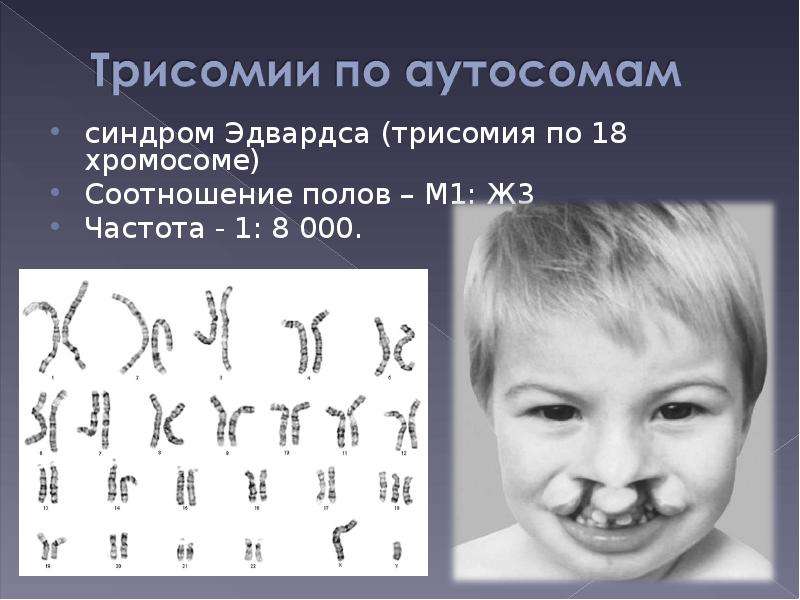 До середины 1900-х годов собрать достаточную информацию об этой патологи не представлялось возможным. Во-первых, для этого необходим был соответствующий уровень развития технологий, который позволил бы обнаружить лишнюю хромосому. Во-вторых, большинство детей умирало в первые дни или недели жизни из-за низкого уровня оказания медицинской помощи. Первое полное описание болезни и ее основной причины (появление лишней 18-й хромосомы) было сделано только в 1960 году врачом Джоном Эдвардом, в честь которого тогда и назвали новую патологию.
До середины 1900-х годов собрать достаточную информацию об этой патологи не представлялось возможным. Во-первых, для этого необходим был соответствующий уровень развития технологий, который позволил бы обнаружить лишнюю хромосому. Во-вторых, большинство детей умирало в первые дни или недели жизни из-за низкого уровня оказания медицинской помощи. Первое полное описание болезни и ее основной причины (появление лишней 18-й хромосомы) было сделано только в 1960 году врачом Джоном Эдвардом, в честь которого тогда и назвали новую патологию.
Дети с синдромом Эдвардса имеют несовместимые с жизнью пороки развития внутренних органов, поэтому основная масса из них не доживает и до одного года. Более половины больных детей умирают в возрасте 2-3 месяцев, а до 12 месяцев доживает лишь около 10% пациентов. В настоящее время разработана государственная программа, включающая 24 стандарта оказания помощи больным с редкими заболеваниями, угрожающими жизни и приводящими к инвалидности.
Ключевые слова: синдром Эдвардса, дети, наблюдение, лечение.
THE EDWARDS SYNDROME AS THE MANIFESTATION OF GENETIC DISEASE IN PEDIATRIC
PRACTICE (clinical case)
E.S. ZAKHAROVA, A.D. LARICOVA
Tula State University, Medical Institute, Boldin Str., 128, Tula, 300028, Russia
Abstract. This article highlights the topic of rare genetic diseases in the practice of a pediatrician using the example of the Edwards syndrome. The authors present an analysis of the clinical case of the detection of this syndrome in a child who was hospitalized in the department for nursing preterm infants. The pediatrician should be aware of the presence of such a formidable disease for timely diagnosis and for choosing tactics for further management of such patients.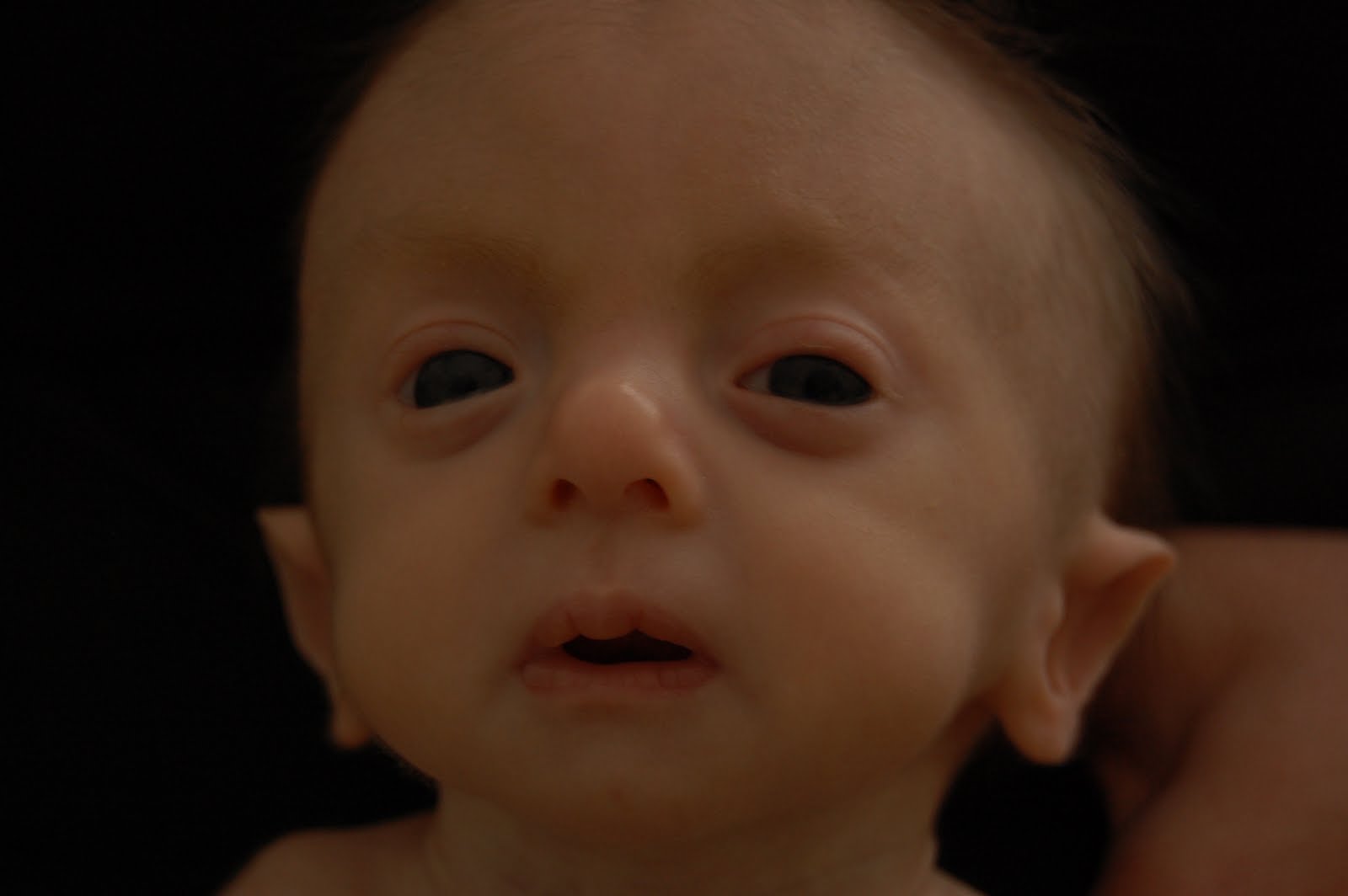 The article presents data on etiology, epidemiology, clinical signs, diagnosis and further treatment tactics for this group of patients.
The article presents data on etiology, epidemiology, clinical signs, diagnosis and further treatment tactics for this group of patients.
The Edwards syndrome (another name — the syndrome of trisomy chromosome 18) is a genetic disease caused by the presence of an additional copy of the eighteenth chromosome, that is, instead of two 18-chromosomes, the patient has three 18 chromosomes. The Edwards syndrome is the second most frequent chromosomal disease after the Down syndrome and it is characterized by a number of defects in the intrauterine development of the child.
The description of the main symptoms of this disease was made at the beginning of the XX century. Until the mid-1900s, it was not possible to gather sufficient information about this pathology. First, this requires an appropriate level of technology development, which would allow us to detect an extra chromosome. Secondly, most children died in the first days or weeks of life because of the low level of medical care. The first complete
The first complete
JOURNAL OF NEW MEDICAL TECHNOLOGIES, eEdition — 2018 — N 2
description of this disease and its main cause (the appearance of an extra 18th chromosome) was made only in 1960 by a physician John Edward, in whose honor the new pathology was named.
Children with the Edwards syndrome have incompatible life-threatening malformations of internal organs, so most of them do not live up to one year. More than half of the sick children die at the age of 2-3 months, and up to 12 months only about 10% of patients survive. Currently, a state program has been developed, which includes 24 standards for the care of patients with rare diseases that threaten life and lead to disability.
Key words: the Edwards syndrome, children, observation, treatment.
Синдром Эдвардса — хромосомное заболевание, обусловленное трисомией по 18-ой хромосоме и сопровождающееся множественными пороками развития. Частота встречаемости синдрома Эдвардса варьирует от 1 на 3000-8000 рождённых [2-4].
Частота встречаемости синдрома Эдвардса варьирует от 1 на 3000-8000 рождённых [2-4].
В патогенезе развития синдрома Эдвардса выступает наличие дополнительной 18-й хромосомы в кариотипе зиготы. Лишняя хромосома у гамет появляется, как правило, из-за нерасхождения хромосом в случае мейотического деления, в результате чего в половой клетке оказывается 24 хромосомы. Если при оплодотворении такой клеткой встречается гамета от противоположного пола, то ими образуется зигота с трисомией.
Существуют следующие вариации: мозаичная трисомия (наличие лишней хромосомы не во всех клетках организма), частичная трисомия (когда присутствует только часть лишней хромосомы), полная трисомия (когда ребенок наследует полную дополнительную копию лишней хромосомы) [4, 6, 7].
Для синдрома Эдвардса характерны: своеобразные фенотипические признаки, аномалии опорно-двигательной системы, аномалии сердечно-сосудистой системы, аномалии пищеварительной системы, аномалии мочеполовой системы, пороки развития ЦНС.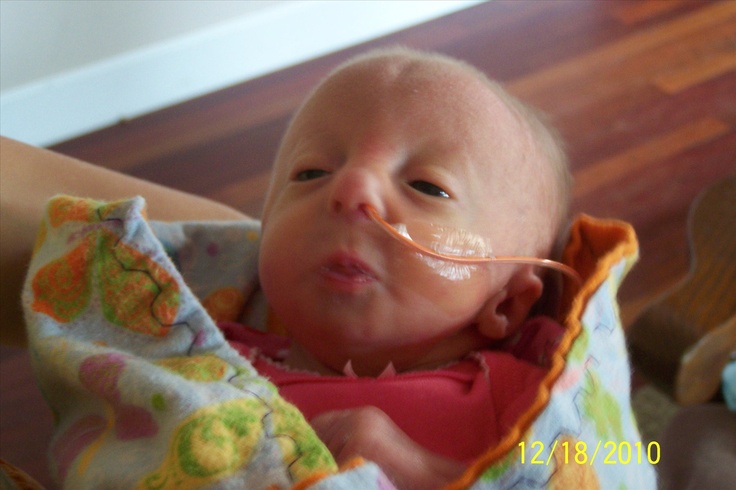
Фенотипические признаки: долихоцефалическая форма черепа, низкий лоб, выступающий затылок, микрогнатия, маленький рот, микрофтальмия, расщелины верхней губы и нёба, эпикантус, птоз, экзофтальм, косоглазие, короткая шея с избыточной кожной складкой, деформации ушных раковин.
Пороки развития опорно-двигательной системы: скрещенные пальцы кистей, укороченная грудина, аномалии ребер, врожденный вывих бедра, косолапость, «стопа-качалка», синдактилия стоп.
Пороки развития сердечно-сосудистой системы: дефекты межжелудочковой и межпредсердной перегородок, коарктация аорты, транспозиция магистральных сосудов, дисплазия клапанов, тетрада Фалло, аномальный дренаж легочных вен, декстракардия.
Пороки развития пищеварительной системы: диафрагмальные, пупочные и паховые грыжи, дивертикул Меккеля, трахеопищеводные свищи, пилоростеноз, атрезия подвздошной кишки и ануса.
Пороки развития мочеполовой системы: подковообразная почка, гидронефроз, дивертикулы мочевого пузыря, гипоспадия и крипторхизм (у мальчиков), двурогая матка, внутриматочная перегородка и гипертрофия клитора (у девочек).
Пороки развития ЦНС: микроцефалия, менингомиелоцеле, гидроцефалия, аномалии Арнольда-Киари, кисты арахноидального сплетения, гипоплазия мозжечка и мозолистого тела [1, 2, 4, 5].
Диагностика включает в себя: УЗИ плода и доплерография маточно-плацентарного кровотока (в ранних сроках беременности можно заподозрить пороки развития головного мозга и конечностей, также наличие обильного количества околоплодной жидкости), анализ крови на сывороточные маркеры (в-субъединицы хорионического гонадотропина (РХГ), а-фетопротеина (АФП), эстриола (ЕЗ), 17-окси прогестерона), инвазивная дородовая диагностика (биопсия хориона, амниоцентез, кордоцентез) с последующим кариотипированием плода.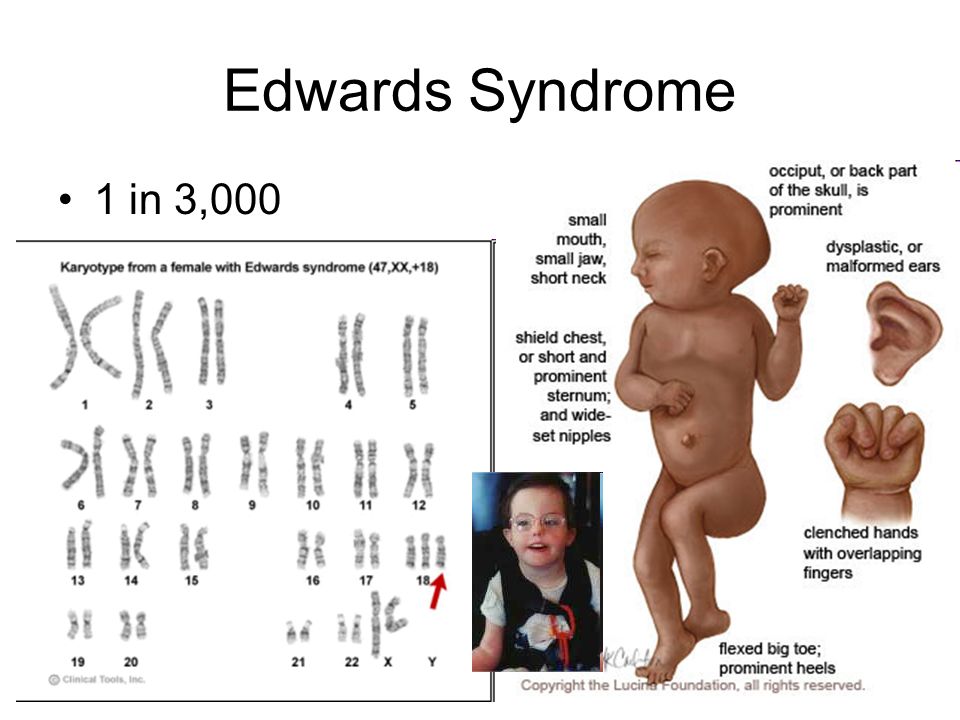
Поскольку в большинстве случаев аномалии развития оказываются несовместимыми с жизнью, лечение детей с синдромом Эдвардса сводится к оказанию симптоматической помощи, направленной на поддержание физиологических функций, продление жизни и улучшение ее качества. Хирургическая коррекция врожденных пороков, как правило, является рискованной и неоправданной [2, 5, 7].
Поскольку дети с синдромом Эдвардса ослаблены и подвержены частой заболеваемости инфекциями мочевыводящих путей, средним отитом, конъюнктивитом, синуситами, пневмонией, они нуждаются в тщательно организованном уходе, полноценном питании, регулярном наблюдении педиатра [1, 2, 8].
Представляем клинический случай синдрома Эдвардса у ребенка в возрасте 1 мес. 25 суток.
Под наблюдением в течение 14 дней находился ребенок в возрасте 1 мес.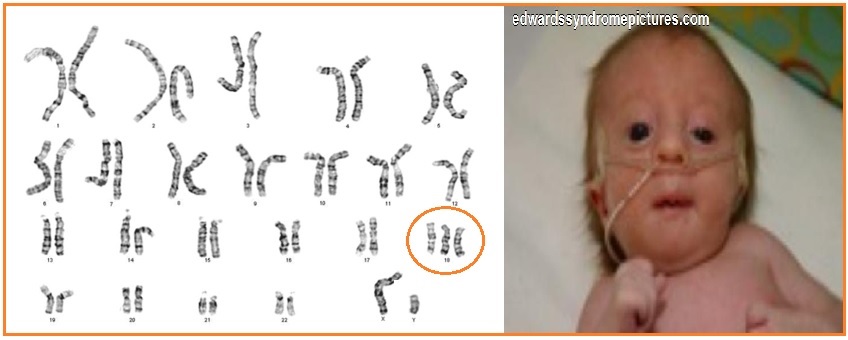 25 суток. Ребенок переведен из ОРИТ в отделение выхаживания недоношенных детей с диагнозом: Множественные врожденные пороки развития: агенезия мозолистого тела. Единственная почка слева. Перинатальное гипокси-чески — ишемическое поражение ЦНС, с-м угнетения. Пневмопатия, ст. разрешения, ДН 0-1 ст. Неона-тальная желтуха. Открытое овальное окно, НК 0 ст. Дисплазия тазобедренных суставов. ЗВУР по гипоксическому типу. Внутриутробное инфицирование.
25 суток. Ребенок переведен из ОРИТ в отделение выхаживания недоношенных детей с диагнозом: Множественные врожденные пороки развития: агенезия мозолистого тела. Единственная почка слева. Перинатальное гипокси-чески — ишемическое поражение ЦНС, с-м угнетения. Пневмопатия, ст. разрешения, ДН 0-1 ст. Неона-тальная желтуха. Открытое овальное окно, НК 0 ст. Дисплазия тазобедренных суставов. ЗВУР по гипоксическому типу. Внутриутробное инфицирование.
Из анамнеза жизни известно: роды 2 срочные в 39 недель, экстренно-оперативным путем кесарева сечения. Поперечное положение плода. Хр. плацентарная недостаточность плода. Анемия беременных. Хр. АГ. Ожирение I ст. При рождении оценка по шкале Апгар на 1 минуте 5 б., на 5 минуте 6
JOURNAL OF NEW MEDICAL TECHNOLOGIES, eEdition — 2018 — N 2
баллов, вес — 1590 г, рост — 44 см. Окружность головы 31 см, груди — 30 см.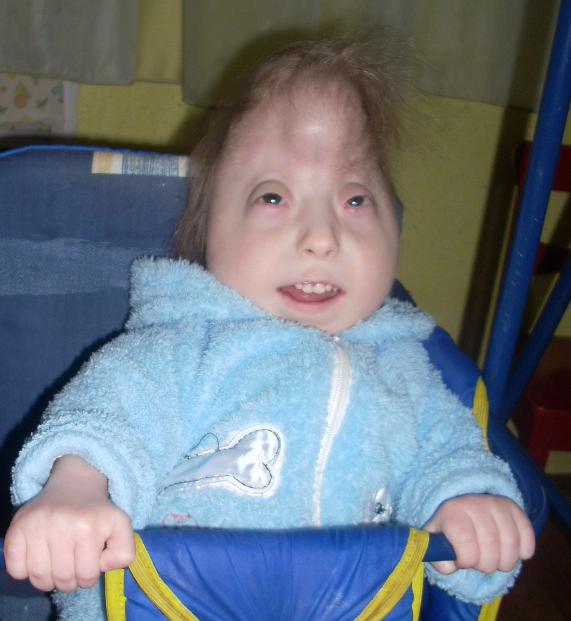 Первичная помощь в род. зале — санация ВДП, рИВЛ через маску 3 минуты + увл. кислородом. Состояние при рождении: тяжелое, обусловленное ДН 1 степени, неврологическим статусом, степенью внутриутробной гипотрофии, внутриутробным инфицированием. По тяжести состояния здоровья ребенок переведен в ОРИТ. Проводимая терапия в ОРИТ: дыхательная поддержка, антибиотикотерапия, инфузионная терапия, фототерапия 2-7 суток, режим кувезный. На фоне проводимого лечения состояние стабилизировалось. Ребенок переведен в отделение выхаживания недоношенных детей с диагнозом:
Первичная помощь в род. зале — санация ВДП, рИВЛ через маску 3 минуты + увл. кислородом. Состояние при рождении: тяжелое, обусловленное ДН 1 степени, неврологическим статусом, степенью внутриутробной гипотрофии, внутриутробным инфицированием. По тяжести состояния здоровья ребенок переведен в ОРИТ. Проводимая терапия в ОРИТ: дыхательная поддержка, антибиотикотерапия, инфузионная терапия, фототерапия 2-7 суток, режим кувезный. На фоне проводимого лечения состояние стабилизировалось. Ребенок переведен в отделение выхаживания недоношенных детей с диагнозом:
Множественные врожденные пороки развития: агенезия мозолистого тела. Единственная почка слева. Перинатальное гипоксически — ишемическое поражение ЦНС, с-м угнетения. Пневмопатия, ст. разрешения, ДН 0-1 ст. Неонатальная желтуха. Открытое овальное окно, НК 0 ст. Дисплазия тазобедренных суставов. ЗВУР по гипоксическому типу. Внутриутробное инфицирование.
Проведено обследование:
Общий анамнез мочи в динамике: от 7. 10: норма. От 15.10, 25.10: дрожжевые грибы
10: норма. От 15.10, 25.10: дрожжевые грибы
Кал на УПФ 15.10: патогенная флора не выявлена.
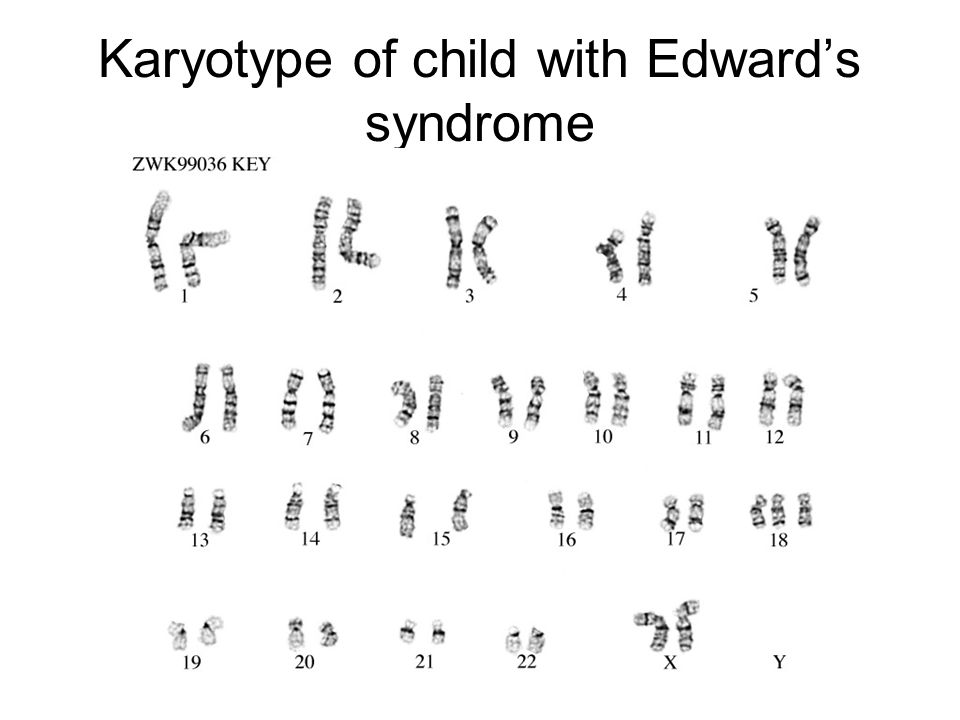
Анализ крови на кариотип от 10.10: Синдром Эдвардса, трисомная форма.
Rg грудной клетки в динамике: от 7.10: легочные поля прозрачные, легочный рисунок усилен в прикорневых зонах. Средостение не смещено.
Rg грудной клетки от 22.10: легочные поля прозрачные, легочный рисунок б/о.
НСГ в динамике: от 7.10: значительные признаки гипоксии, незрелости. Аномалия строения головного мозга: агенезия мозолистого тела. Дилатация тел и задних рогов боковых желудочков I-II ст. Псевдокисты: ПВК I-II ст. слева и сосудистого сплетения справа. Дилатация цистерны мозга. НСГ от 21.10: без динамики.
ЭХО-КГ от 7. 10: ООО 3 мм. Аномалия строения АоК
10: ООО 3 мм. Аномалия строения АоК
УЗИ брюшной полости от 10.10: единственная галетообразная почка слева. Деформация желчного пузыря.
УЗИ тазобедренных суставов от 13.10: умеренная степень дисплазии.
ЭКГ от 8.10: ритм синусовый, правильный. ЭОС — полувертикальное положение.
Окулист от 13.10: преретинопатия (высокий риск прогрессирования).
Невролог от 14.10: гипоксически-ишемическое поражение ЦНС
Ортопед от 25.10: дисплазия тазобедренных суставов.
Генетик от 10.10: Синдром Эдвардса, трисомная форма.
Лечение, проводимое в отделении выхаживания недоношенных детей.
— Антибактериальная терапия (Цефомакс 80 мг в/в 2 раза в день)
— Профилактика и лечение капиллярных кровотечений (Этамзилат 2,5% 0,3 мл в/в 2 раза в день)
— Улучшение метаболизма (Элькар 30% 5 кап.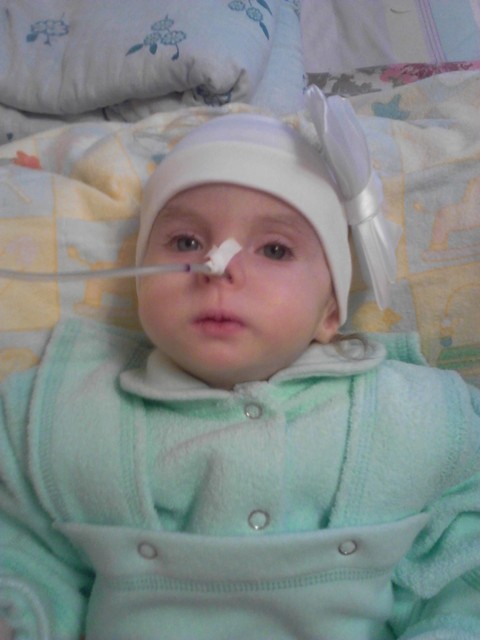 per os 2 раза в день)
per os 2 раза в день)
— Профилактика дисбактериоза (Бифидумбактерин 2,5 дозы per os 2 раза в день)
— Витамины А (1 кап. 1 раз в день per os) и Е(1 кап. 1 раз в день per os)
— Эмоксипин 1 % (1 кап. 6 раз/час) 1 раз в сутки в глаза.
После отделения выхаживания недоношенных детей при достижении массы тела 2500 гр. ребенок был переведен в областной дом ребенка.
Большинство детей, которые родились с синдромом Эдвардса, не доживают до первого года своей жизни. Средняя продолжительность жизни для половины детей, рожденных с этим синдромом, менее чем два месяца. От девяноста до девяноста пяти процентов из этих детей умирает, не дожив до своего первого дня рождения. От пяти до десяти процентов детей, которые выжили в первый год, испытывают серьезные отклонениями в развитии.
Разработана государственная программа по профилактике орфанных заболеваний (закон РФ от 21 ноября 2011 г. № 323-ФЗ «Об основах охраны здоровья граждан в Российской Федерации»), которая содержит следующие пункты: планирование беременности, контроль за состоянием здоровья женщины, витаминотерапия в течение беременности, регулярное наблюдение беременной в условиях женской консультации.
Литература
1. Вахарловский В.Г. Генетика в практике педиатра. Руководство для врачей. Изд-во СПбГПМА, 2009. 286 с.
2. Вахарловский В.Г., Горбунова В.Н. Клиническая генетика. СПб.: Изд-во СПбГПМА, 2010. №38.
3. Иванов В.И. Генетика: учебник для вузов / под ред. акад. Иванова В.И. М.: ИКЦ «Академкнига», 2009. 638 с.
4. Козлова С.И., Демикова Н.С. Наследственные синдромы и медико-генетическое консультирование. М., 2010. 448 с.
Козлова С.И., Демикова Н.С. Наследственные синдромы и медико-генетическое консультирование. М., 2010. 448 с.
ВЕСТНИК НОВЫХ МЕДИЦИНСКИХ ТЕХНОЛОГИЙ, электронный журнал — 2018 — N 2 JOURNAL OF NEW MEDICAL TECHNOLOGIES, eEdition — 2018 — N 2
5. Курзина E.A. Роль наследственных заболеваний у детей с перинатальной патологией // Вестник Педиатрической академии. 2009. № 5. С. 96-101.
6. Новиков П.В. Семиотика наследственных болезней у детей (симптом-синдром-болезнь). М.: Триада-Х, 2009. 432 с .
7. Таточенко В.К. Педиатру на каждый день. Справочник по диагностике и лечению. Издание восьмое, дополненное. М., 2016. 271 с.
8. Шабалов Н.П. Справочник педиатра. 3-е издание П., 2015. 577 с.
References
1. Vakharlovskiy VG. Genetika v praktike pediatra [Genetics in the practice of pediatrician. A guide for physicians]. Rukovodstvo dlya vrachey. Izd-vo SPbGPMA; 2009. Russian.
Vakharlovskiy VG. Genetika v praktike pediatra [Genetics in the practice of pediatrician. A guide for physicians]. Rukovodstvo dlya vrachey. Izd-vo SPbGPMA; 2009. Russian.
2. Vakharlovskiy VG, Gorbunova VN. Klinicheskaya genetika [Clinical genetics]. Sankt-Peterburg: Izd-vo SPbGPMA; 2010. Russian.
3. Ivanov VI. Genetika: uchebnik dlya vuzov [Genetics: textbook for universities]. Pod red. akad. Ivanova VI. Moscow: IKTs «Akademkniga»; 2009. Russian.
4. Kozlova SI, Demikova NS. Nasledstvennye sindromy i mediko-geneticheskoe konsul’tirovanie [Hereditary syndromes and medical genetic counseling]. Moscow; 2010. Russian.
5. Kurzina EA. Rol’ nasledstvennykh zabolevaniy u detey s perinatal’noy patologiey [the role of hereditary diseases in children with perinatal pathology]. Vestnik Pediatricheskoy akademii. 2009;5:96-101. Russian.
2009;5:96-101. Russian.
6. Novikov PV. Semiotika nasledstvennykh bolezney u detey (simptom-sindrom-bolezn’) [Semiotics of hereditary diseases in children (symptom-syndrome-disease)]. Moscow: Triada-Kh; 2009. Russian.
7. Tatochenko VK. Pediatru na kazhdyy den’. Spravochnik po diagnostike i lecheniyu. [the Pediatrician every day. Guide to diagnosis and treatment] Izdanie vos’moe, dopolnennoe. Moscow; 2016. Russian.
8. Shabalov NP. Spravochnik pediatra [Handbook of pediatrician]. 3-e izdanie P.; 2015. Russian.
Библиографическая ссылка:
Захарова Е.С., Ларикова А.Д.Синдром Эдвардса как проявление генетического заболевания в педиатрической практике (клинический случай) // Вестник новых медицинских технологий. Электронное издание. 2018. №2. Публикация 1-3. URL: http://www.medtsu.tula.ru/VNMT/Bulletin/E2018-2/1-3.pdf (дата обращения: 15.03.2018). DOI: 10.24411/2075-4094-2018-16008.
2018. №2. Публикация 1-3. URL: http://www.medtsu.tula.ru/VNMT/Bulletin/E2018-2/1-3.pdf (дата обращения: 15.03.2018). DOI: 10.24411/2075-4094-2018-16008.
Редкий нетипичный случай синдрома Эдвардса Текст научной статьи по специальности «Клиническая медицина»
УДК 616.8-053
М.М. Лепесова, Д.Т. Отегенова, А.М. Курмантай*
Казахский медицинский универси те т непрерывного образования,
г. Алматы, Казахстан
РЕДКИЙ НЕТИПИЧНЫЙ СЛУЧАЙ СИНДРОМА ЭДВАРДСА
АННОТАЦИЯ
Рассмотрен клинический случай у девочки 10 мес. с неклассическим проявлением фенотипа синдрома Эдвардса, что вызвало сомнение при постановке диагноза. Синдром Эдвардса или синдром трисомии 18 — это общее хромосомное расстройство, обусловленное наличием во всех клетках организма добавочной хромосомы 18. Такие дети рождаются с задержкой внутриутробного развития. Клинические симптомы заболевания: гипогнотия, выраженная задержка психического и моторного развития, множественные врожденные пороки: костно-мышечной, сердечно-сосудистой, желудочно-кишечной и центральной нервной систем. Также отмечаются изменения со стороны органов зрения: помутнение корнеальных оболочек, катаракты, птоз, микрофтальмия. Отличительные признаки: сжатые кулаки с перекрытием пальцев, короткая грудина, кожный узор в виде дуг на большинстве пальцев.. На примере клинического случая, описанного в статье, отмечается полиморфизм синдрома Эдвардса: у данного ребенка отсутствовали типичные изменения костно-мышечной системы. Только с помощью генетического исследования удалось установить у пациента хромосомное расстройство синдром Эдвардса. Ключевые слова: синдром Эдвардса, трисомия 18, ребенок.
Синдром Эдвардса или синдром трисомии 18 — это общее хромосомное расстройство, обусловленное наличием во всех клетках организма добавочной хромосомы 18. Такие дети рождаются с задержкой внутриутробного развития. Клинические симптомы заболевания: гипогнотия, выраженная задержка психического и моторного развития, множественные врожденные пороки: костно-мышечной, сердечно-сосудистой, желудочно-кишечной и центральной нервной систем. Также отмечаются изменения со стороны органов зрения: помутнение корнеальных оболочек, катаракты, птоз, микрофтальмия. Отличительные признаки: сжатые кулаки с перекрытием пальцев, короткая грудина, кожный узор в виде дуг на большинстве пальцев.. На примере клинического случая, описанного в статье, отмечается полиморфизм синдрома Эдвардса: у данного ребенка отсутствовали типичные изменения костно-мышечной системы. Только с помощью генетического исследования удалось установить у пациента хромосомное расстройство синдром Эдвардса. Ключевые слова: синдром Эдвардса, трисомия 18, ребенок.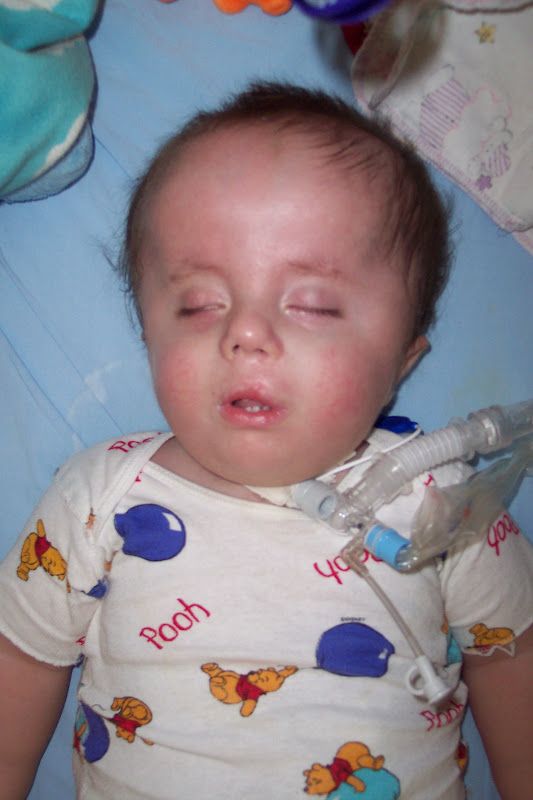
Синдром трисомии 18 (синдром Эдвардса) -хромосомное расстройство, обусловленное наличием добавочной хромосомы 18. Этот синдром был впервые описан Джоном Эдвар-дсом как отдельная хромосомная болезнь в 1960 г. По частоте занимает второе место среди синдромов множественных пороков развития, встречается у 1 из 3000 новорожденных. У девочек встречаемость в 3 раза больше, чем у мальчиков. Беременность может проходить на фоне многоводия, гиподинамией плода в утробе матери и часто роды наступают на поздних сроках. 60 % детей с данной патологией умирают в утробе матери. Продолжительность жизни составляет для мальчиков 2 месяца, а для девочек 10 месяцев [4]. Различают три вида хромосомного дефекта данного заболевания:
1) Полная трисомия наличие во всех клетках организма дополнительной 18-й хромосомы. Данная форма заболевания встречается в 90 % случаев и является наиболее тяжелой.
2) Частичная трисомия 18 является относительно редким (не более 3 % всех случаев синдрома Эдвардса, который характеризуется наличием фрагмента дополнительной хромосомы. Причинами могут быть нарушение в делении генетического материала. У таких де-
Причинами могут быть нарушение в делении генетического материала. У таких де-
тей прогноз лучше, чем у детей с полной формой, однако исход остается неблагоприятный.
3) Мозаичная форма синдрома Эдвардса встречается в 5-7 % случаев. Нарушение в структуре хромосомы видно уже после слияния сперматозоида и яйцеклетки. Они имели нормальный кариотип и несли по одной хромосоме каждого вида. После слияния сформировалась клетка с нормальной формулой 46,XX или 46,ХУ. В процессе мейоза клетки произошёл дефект. При удвоении генетического материала один из фрагментов получил дополнительную 18-ю хромосому. Таким образом, сформировался дефект, часть клеток которого имеет нормальный кариотип (например, 46,XX), а часть — пораженный (47,XX, 18+). В конечном итоге все клетки организма представляют собой своеобразную мозаику [5]. У таких детей задержка пренатального развития. Общие признаки: слабое шевеление плода, тихий крик, несвоевременные роды, многоводие, маленькая плацента, средний вес при рождениях 2340 г. , умственная отсталость, тугоухость. Голова: долихоцефалической формы, узкий лоб, короткие глазные щели, микростомия, узкое высокое небо, низко расположенные ушные раковины, аномалии развития неба и верхней губы, нижняя микрогнатия [6]. Кисти и стопы:
, умственная отсталость, тугоухость. Голова: долихоцефалической формы, узкий лоб, короткие глазные щели, микростомия, узкое высокое небо, низко расположенные ушные раковины, аномалии развития неба и верхней губы, нижняя микрогнатия [6]. Кисти и стопы:
*Курмантай А.М. [email protected]
25
] КАЗАХСКИЙ МЕДИЦИНСКИЙ IУНИВЕРСИТЕТ НЕПРЕРЫВНОГО ОБРАЗОВАНИЯ
Вестник АГИУВ № 2, 2016
сжатые кулаки с частым перекрыванием вторым пальцем третьего, а пятым четвертого, отсутствие дистальной сгибательной борозды на V пальце рук, гипоплазия ногтей, часто переразогнутый 1 палец на ногах, ненормальная пропорция в длине пальцев ног. Грудная клетка: короткая грудина с уменьшенным количеством ядер окостенения, гипоплазия сосков. Живот: пупочная грыжа, паховая грыжа, расхождения прямых мышц живота.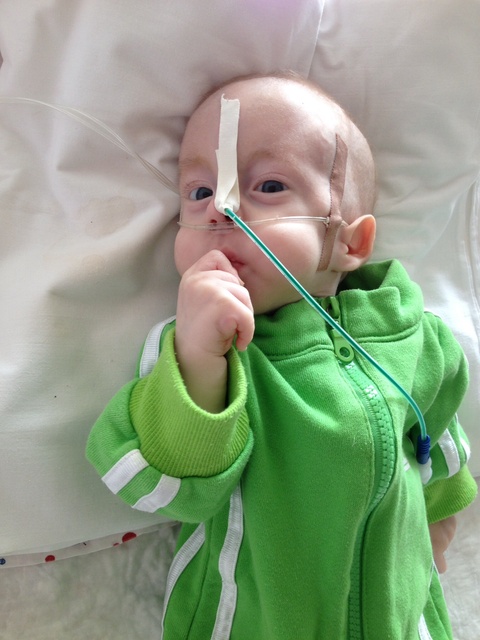 Таз: маленький, неполное отведение бедер. Половые органы: у мальчиков — крипторхизм. Кожа: складчатая, с выраженным рисунком, легкое оволосение лба и спины. Сердце: дефекты межпред-сердной и межжелудочковой перегородки, открытый артериальный проток. Голова: широкие роднички, микроцефалия, эпикант, птоз, помутнение роговицы, расщелина верхней губы и неба. Кисти и стопы: локтевая и лучевая косорукость, гипоплазия или аплазия 1 пальца кисти, конско-варусная стопа, стопа-качалка, синдактилия II и III пальцев. Грудная клетка: короткая и широкая, возможен гипертелоризм сосков. Половые органы: у девочек гипоплазия больших половых губ и гипертрофия половых губ. Прямая кишка: эктопия или воронкообразная втянутость заднего прохода. Легкие: нарушения легочной сегментации, гипоплазия легких, аплазия правого легкого. Диафрагма: врожденное недоразвитие мышечной ткани, возможной релаксацией диафрагмы. Живот: дивертикул Меккеля, добавочная селезенка, добавочная поджелудочная железа, грыжа пупочного канатика, незавершенный поворот кишечника.
Таз: маленький, неполное отведение бедер. Половые органы: у мальчиков — крипторхизм. Кожа: складчатая, с выраженным рисунком, легкое оволосение лба и спины. Сердце: дефекты межпред-сердной и межжелудочковой перегородки, открытый артериальный проток. Голова: широкие роднички, микроцефалия, эпикант, птоз, помутнение роговицы, расщелина верхней губы и неба. Кисти и стопы: локтевая и лучевая косорукость, гипоплазия или аплазия 1 пальца кисти, конско-варусная стопа, стопа-качалка, синдактилия II и III пальцев. Грудная клетка: короткая и широкая, возможен гипертелоризм сосков. Половые органы: у девочек гипоплазия больших половых губ и гипертрофия половых губ. Прямая кишка: эктопия или воронкообразная втянутость заднего прохода. Легкие: нарушения легочной сегментации, гипоплазия легких, аплазия правого легкого. Диафрагма: врожденное недоразвитие мышечной ткани, возможной релаксацией диафрагмы. Живот: дивертикул Меккеля, добавочная селезенка, добавочная поджелудочная железа, грыжа пупочного канатика, незавершенный поворот кишечника. Почки: подковообразная почка, дистопия почек, удвоение мочеточника, гидронефроз, поликистоз почек и кишечника. ЦНС: парез мимических мышц, слабая миелинизация, микрогирия, гипоплазия мозжечка, гипоплазия мозолистого тела, гидроцефалия. Прочее: ге-мангиомы, гипоплазия вилочковой железы, тра-хеопищеводные свищи, тромбоцитопения. Множественные пороки развития обуславливают раннюю смертность детей с данной патологией. В связи с вышесказанным, прогноз заболевания является неблагоприятным: лишь 5-10 % больных живут до 1 года и менее 1 % — до 10 лет и старше [7,8]. В течении новорожденнос-ти у детей часто наблюдаются нарушения мозгового кровообращения, кишечная непроходимость, что приводит к смерти этих детей. Дети более старшего возраста умирают обыч-
Почки: подковообразная почка, дистопия почек, удвоение мочеточника, гидронефроз, поликистоз почек и кишечника. ЦНС: парез мимических мышц, слабая миелинизация, микрогирия, гипоплазия мозжечка, гипоплазия мозолистого тела, гидроцефалия. Прочее: ге-мангиомы, гипоплазия вилочковой железы, тра-хеопищеводные свищи, тромбоцитопения. Множественные пороки развития обуславливают раннюю смертность детей с данной патологией. В связи с вышесказанным, прогноз заболевания является неблагоприятным: лишь 5-10 % больных живут до 1 года и менее 1 % — до 10 лет и старше [7,8]. В течении новорожденнос-ти у детей часто наблюдаются нарушения мозгового кровообращения, кишечная непроходимость, что приводит к смерти этих детей. Дети более старшего возраста умирают обыч-
но от пневмонии на фоне недостаточности сердечно-сосудистой системы или от инфекций мочевыводящих путей [9]. Клиническая и в некоторых случаях патологоанатомическая дифференциальная диагностика данной патологии вызывает затруднение, поэтому всем в обязательном порядке надо провести цитоге-нетическое исследование.
На примере клинического случая данного синдрома у ребенка 10 месяцев рассмотрим некоторые аспекты данной патологии: девочка А. 2015 г. р., впервые поступила в отделение неврологии младшего возраста № 2 Городской детской клинической больницы (ГДКБ) г. Ал-маты. Жалобы матери при поступлении на задержку моторного развития (не сидит, голову не держит, не ползает). Анамнез жизни: ребенок от второй беременности, вторых самостоятельных родов. От первой беременности ребенок здоровый. Данная беременность протекала на фоне ОРВИ в первом триместре, анемии легкой степени в течении всей беременности. На 19-й неделе беременности по УЗИ плода диагностирован МВПР у плода. Врожденный порок сердца: дефект межжелудочковой (ДМЖП). ВПР мочеполовой системы. Агенезия правой почки. ЭГМХА, киста сосудистого сплетения головного мозга. Родители от прерывания беременности отказались. Роды самостоятельные, протекли без особенностей, в сроке 41 недели, родилась девочка весом 2412 г, ростом 46 см. Оценили по шкале Апгар на 6-7 баллов. Состояние ребенка при рождении было тяжелым за счет дыхательной недостаточности. Первые 3 дня жизни ребенок находился в отделении детской реанимации с диагнозом: респираторный дистресс-синдром, ДН 3 степени. Перинатальное поражение ЦНС, синдром угнетения. Задержка внутриутробного развития. С 3 дня жизни ребенок был переведен на второй этап выхаживания, где получал дальнейшее лечение. На 16-й день выписались домой, под наблюдением педиатра по месту жительства. Через неделю и в дальнейшем 3 раза получала стационарное лечение в детской инфекционной больнице с диагнозом: «острая респираторная вирусная инфекция. Острый обструктивный бронхит, тяжелая форма». Сопутствующий диагноз: ДМЖП, ОАП, ООО. Диафрагмальная грыжа слева. БЭН 2ст. Гиперплазия вилочковой железы 2 см». На 10-м месяце жизни ребенок
Оценили по шкале Апгар на 6-7 баллов. Состояние ребенка при рождении было тяжелым за счет дыхательной недостаточности. Первые 3 дня жизни ребенок находился в отделении детской реанимации с диагнозом: респираторный дистресс-синдром, ДН 3 степени. Перинатальное поражение ЦНС, синдром угнетения. Задержка внутриутробного развития. С 3 дня жизни ребенок был переведен на второй этап выхаживания, где получал дальнейшее лечение. На 16-й день выписались домой, под наблюдением педиатра по месту жительства. Через неделю и в дальнейшем 3 раза получала стационарное лечение в детской инфекционной больнице с диагнозом: «острая респираторная вирусная инфекция. Острый обструктивный бронхит, тяжелая форма». Сопутствующий диагноз: ДМЖП, ОАП, ООО. Диафрагмальная грыжа слева. БЭН 2ст. Гиперплазия вилочковой железы 2 см». На 10-м месяце жизни ребенок
поступает в отделение неврологии младшего возраста. При поступлении общее состояние ребенка средней тяжести за счет неврологических признаков.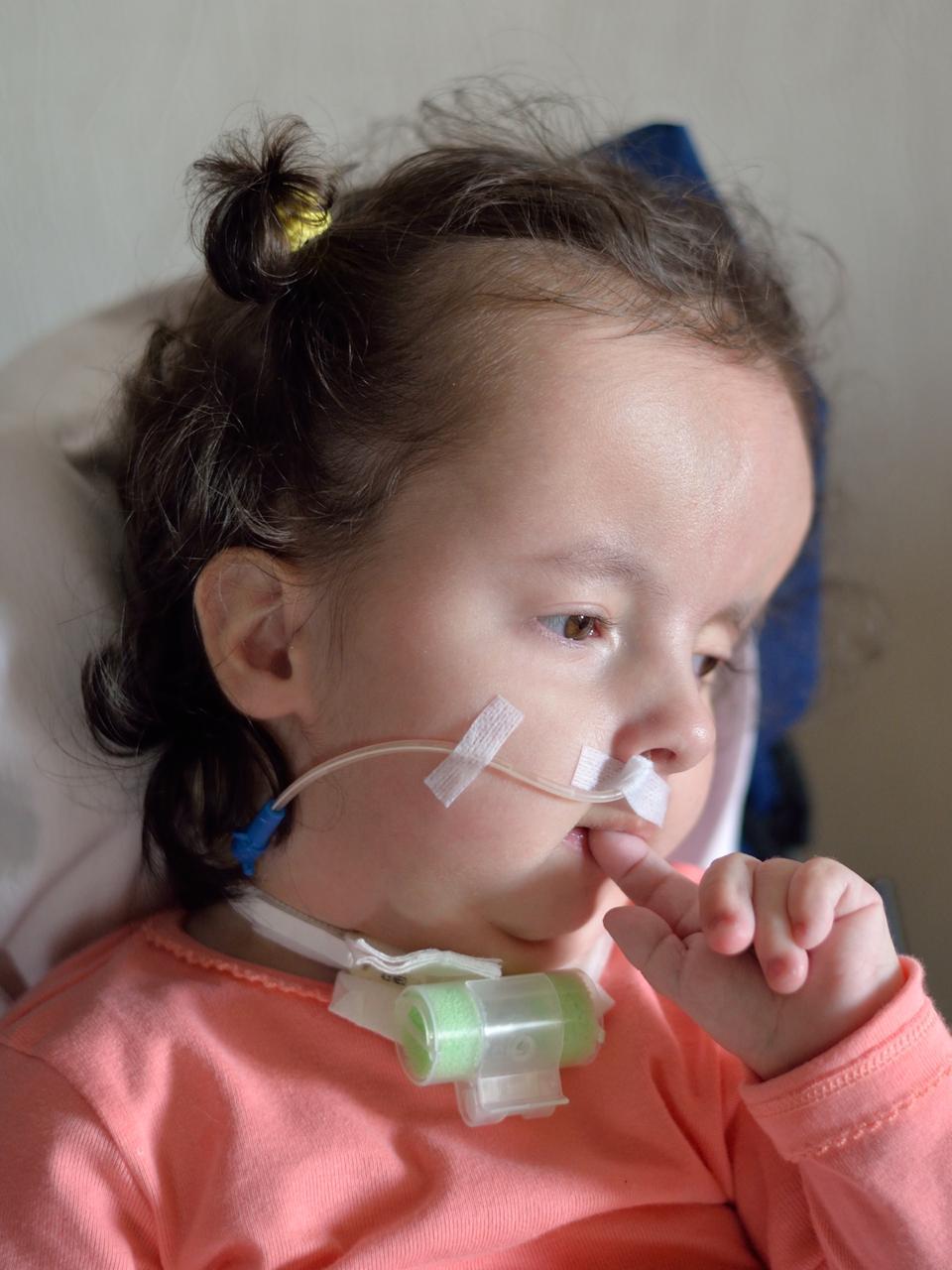 В соматическом статусе: физическое развитие ниже среднего, вес 4,300 кг, окружность головы 39 см (микроцефалия). Кожа бледной окраски с мраморным оттенком. Подкожно-жировой слой развит недостаточно. При первичном осмотре ребенка определяются: широкая переносица, низко расположенные ушные раковины, отсутствует козелок уха, редкий рост волос и аномальный рост ресниц, эпикант, микрофтальмия, птоз, недоразвитие нижней челюсти, стопа «качалка», маленький лоб и рот. Движения в тазобедренном суставе не в полном объеме. Грудная клетка при осмотре вздутая, выраженное слева. Со стороны дыхательной и сердечно-сосу-дистой системы без особенностей. Имеются патология строения половых органов: гипертрофированный клитор и недоразвитые половые губы. Стул и мочеиспускание без особенностей (со слов мамы). Неврологический статус: в сознании, вялый. Эмоциональная сфера снижена, лицо гипомимичное, в окружающем не ориентируется. Менингеальные знаки отрицательные. Со стороны 12 пар черепных нервов: сходящееся косоглазие.
В соматическом статусе: физическое развитие ниже среднего, вес 4,300 кг, окружность головы 39 см (микроцефалия). Кожа бледной окраски с мраморным оттенком. Подкожно-жировой слой развит недостаточно. При первичном осмотре ребенка определяются: широкая переносица, низко расположенные ушные раковины, отсутствует козелок уха, редкий рост волос и аномальный рост ресниц, эпикант, микрофтальмия, птоз, недоразвитие нижней челюсти, стопа «качалка», маленький лоб и рот. Движения в тазобедренном суставе не в полном объеме. Грудная клетка при осмотре вздутая, выраженное слева. Со стороны дыхательной и сердечно-сосу-дистой системы без особенностей. Имеются патология строения половых органов: гипертрофированный клитор и недоразвитые половые губы. Стул и мочеиспускание без особенностей (со слов мамы). Неврологический статус: в сознании, вялый. Эмоциональная сфера снижена, лицо гипомимичное, в окружающем не ориентируется. Менингеальные знаки отрицательные. Со стороны 12 пар черепных нервов: сходящееся косоглазие.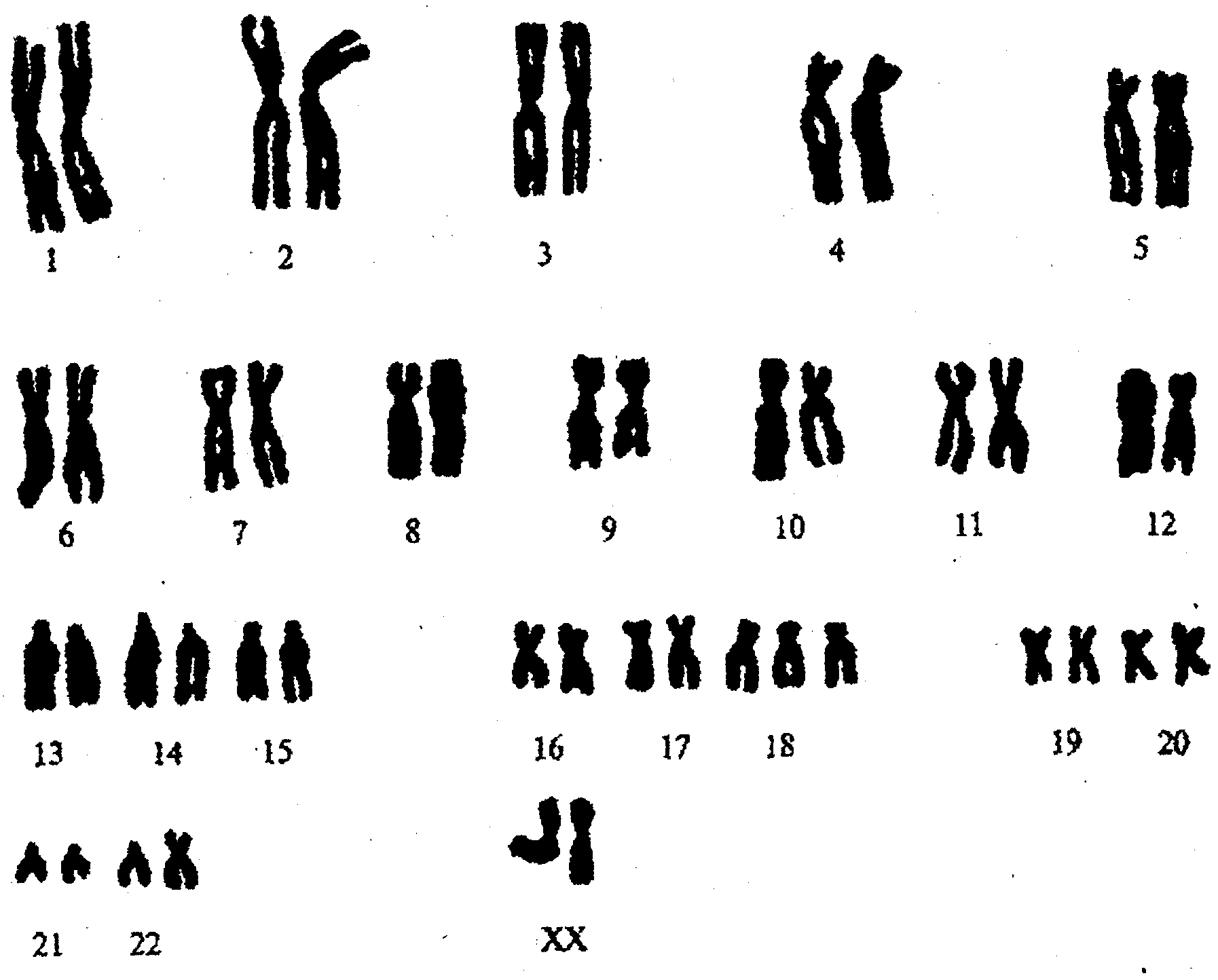 Фотореакции сохранена. В обоих глазах имеется частичная катаракта. За предметами не следит. Слух не нарушен. Нистагм не определяется. При употреблении еды периодически поперхивается. Голос слабый, изредка издает монотонные звуки. Фасцикуляции и фибрилляции отсутствуют. В двигательной сфере: мышечный тонус дистоничен. Контрактуры нет. Сухожильные рефлексы не вызываются, 13=8. Голову не удерживает, не сидит, не переворачивается. Учитывая вышеизложенные данные, нами был поставлен предварительный диагноз: наследственный синдром. Задержка психомоторного развития. БЭН 2- степени. Дисплазия тазобедренного сустава. Лабораторные и инструментальные данные: Общий анализ крови и общий анализ мочи: без изменения.
Фотореакции сохранена. В обоих глазах имеется частичная катаракта. За предметами не следит. Слух не нарушен. Нистагм не определяется. При употреблении еды периодически поперхивается. Голос слабый, изредка издает монотонные звуки. Фасцикуляции и фибрилляции отсутствуют. В двигательной сфере: мышечный тонус дистоничен. Контрактуры нет. Сухожильные рефлексы не вызываются, 13=8. Голову не удерживает, не сидит, не переворачивается. Учитывая вышеизложенные данные, нами был поставлен предварительный диагноз: наследственный синдром. Задержка психомоторного развития. БЭН 2- степени. Дисплазия тазобедренного сустава. Лабораторные и инструментальные данные: Общий анализ крови и общий анализ мочи: без изменения.
На ЭКГ: синусовая тахикардия ЧСС 166-ударов в минуту. Нормальное положение ЭОС. Частичная блокада правой ножки пучка Гисса. Повышена электрическая активность правого желудочка.
На УЗИ ОБП: диффузные изменения пе-
чени. ВПР МВС. Дистопия правой почки, по-яснично-крестцовая?
ВПР МВС. Дистопия правой почки, по-яснично-крестцовая?
На КТ головного мозга: ретроцеребелляр-ная ликворная киста.
На КТ органов грудной клетки: врожденная диафрагмальная грыжа, истинная слева. Воспалительные изменения паренхимы легких. КТ картина характерна для двухсторонней полисегментарной пневмоний.
Рентгенография тазобедренного сустава: дисплазия тазобедренных суставов.
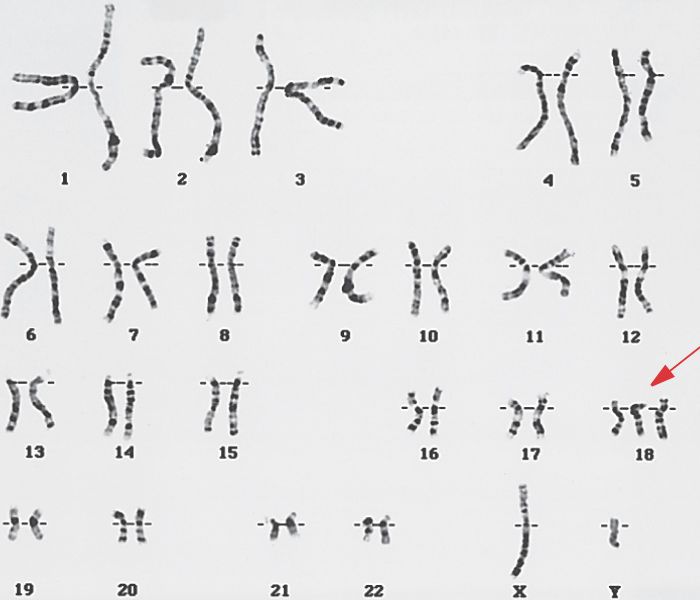
При кариотипировании обнаружено 47 хромосом, трисомия по 18-й хромосоме, что позволяет подтвердить диагноз наследственной патологии. Во время пребывания в стационаре у девочки ухудшилось состояние в связи с присоединением острой респираторной инфекции, осложнившаяся двухсторонней пневмонией. Получала антибактериальную терапию. После стабилизации состояния, на 13-й день госпитализации была выписана домой под наблюдением участкового педиатра и детского невролога по месту жительства с соответствующими рекомендациями.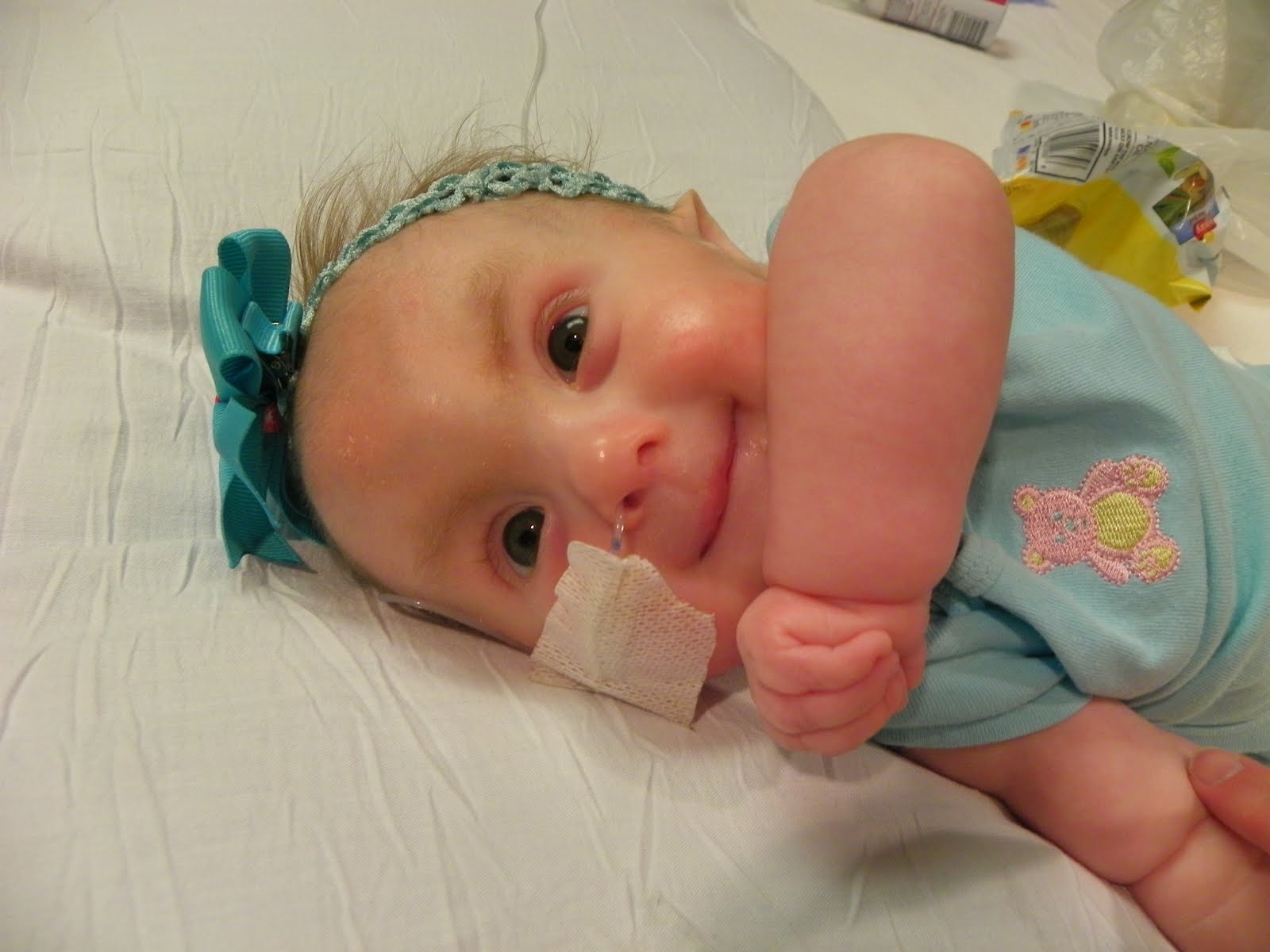 Прогноз в данном случае неблагоприятный. Даже самые современные методы лечения и ухода могут лишь поддерживать жизнь ребенка. Ни в каких протоколах нет единых рекомендаций по уходу за детьми с данной хромосомной мутацией. Подводя итоги, хотелось бы подчеркнуть, что приведенное клиническое наблюдение демонстрирует наименее изученный вариант аномалии половых хромосом: синдром Эдвардса. Дети с задержкой нервно-психического развития, часто болеющие и имеющие стигмы дисэмбрио-генеза нуждаются в обязательном генетическом обследовании, чтобы диагностировать наследственное хромосомное заболевание даже при отсутствии классических признаков.
Прогноз в данном случае неблагоприятный. Даже самые современные методы лечения и ухода могут лишь поддерживать жизнь ребенка. Ни в каких протоколах нет единых рекомендаций по уходу за детьми с данной хромосомной мутацией. Подводя итоги, хотелось бы подчеркнуть, что приведенное клиническое наблюдение демонстрирует наименее изученный вариант аномалии половых хромосом: синдром Эдвардса. Дети с задержкой нервно-психического развития, часто болеющие и имеющие стигмы дисэмбрио-генеза нуждаются в обязательном генетическом обследовании, чтобы диагностировать наследственное хромосомное заболевание даже при отсутствии классических признаков.
Выводы. На примере клинического случая предоставлено редкое хромосомное заболевание — синдром Эдвардса. У данного пациента не было типичных проявлений: видимых изменении костно-мышечной системы, что характерно при данной патологии. Это вызвало затруднения в постановке диагноза. Исходя из этого, делаем вывод: что нетипичное проявление хромосомного заболевания не отрицает его наличия и для уточнения диагноза обязательно надо провести генетическое исследование.
1 Вахарловский В.Г., Горбунова В.Н. Клиническая генетика. — СПб., Изд-во СПбГПМА. -2007.-№38.
2 Edwards J. A new trisomic syndrome. I I Lancet, 1960. — 787 p.
3 Иванов В. И. и др. Генетика: учебник для вузов / под ред. акад. В. И. Иванова. — М.: ИКЦ «Академкнига», 2006. — 638 с.
4 Козлова С.И., Демикова Н.С. Наследственные синдромы и медико-генетическое консультирование. — М., 2007. — 448 с.
5 HallJ. G. Chromosomal clinical anomalies. — Philadelphia-London , 2000. — 325 с.
6 Курзина E.A. и др. Роль наследственных заболеваний у детей с перинатальной патологией // Вестник Педиатрической академии. — 2007. — № 5. — С. 96-101.
7 Новиков П. В. Семиотика наследственных болезней у детей (симптом-синдром-болезнь). -М.: Триада-Х, 2009. — 432 с .
В. Семиотика наследственных болезней у детей (симптом-синдром-болезнь). -М.: Триада-Х, 2009. — 432 с .
8 Gessner В. D. Reasons for trisomy 13 and 18 births despite the availability of prenatal diagnosis and pregnancy termination.Early Human Development. — 2003. — № (73. — P. 53-60.
9 Munne S. et al. Increased rate of aneuploid embryos in young women with previous aneuploid conceptions.Prenatal Diagnosis. — 2004. — № 24. — P. 638-643.
TYI/IIH
Буя макалада диагноз коюда киындьщ тугызган Эдварде синдромыныц классикальщ емес фенотигп орын алган 10 айльщ кыздыц клиникальщ жагдайын карастырамыз. Эдварде синдромы немесе 18 трисо-мия синдромы — бул барльщ агза жасушасында косымша 18 хромосома болуымен сипатталатын жалпы хромосомальщ ауытку. Мундай балалар KypcaKiLumiK дамудыц кемшттмен туылады.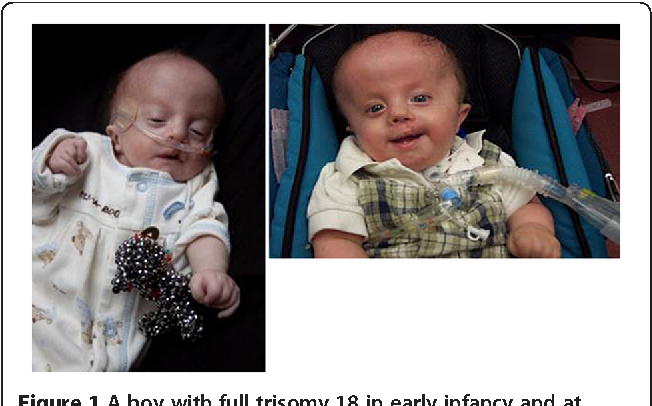 Аурудыц клиникалык симптомдары: гипогнотия, туа б1ткен психика жэне кимыл-козгалыс дамуыныц ерескел тежелу1, кептеген туа бггкен акаулар: суйек-булшьщет, журек-кан-тамыр, асказанчшек жэне орталык жуйке жуйелершщ. Сондай-ак, керу мушеа жагынан езгер1стер болады: касак кабыктьщ мелд1рл1пнщ жогалуы, катаракта, птоз, микро-фтальмия. Айрьщша 6enrmepi: саусактарын жабарльщтай жудырыгыныц туюл1 болуы, TeciHiK кыскалыгы, б1рнеше саусагында дога T8pi3fli TepiniK суреттщ болуы. Макалада керсеттген клиникальщ жагдай мысалын-да Эдварде синдромыньщ полиморфтылыгы байкалады: бул балада суйек-булшьщет жуйесшщ тон 93repi-CTepi табылган жок. Наукаска Эдварде синдромы сиякты хромосомальщ ауытку генетикальщ зерттеу нэти-жеа кемег1мен гана койылды.
Аурудыц клиникалык симптомдары: гипогнотия, туа б1ткен психика жэне кимыл-козгалыс дамуыныц ерескел тежелу1, кептеген туа бггкен акаулар: суйек-булшьщет, журек-кан-тамыр, асказанчшек жэне орталык жуйке жуйелершщ. Сондай-ак, керу мушеа жагынан езгер1стер болады: касак кабыктьщ мелд1рл1пнщ жогалуы, катаракта, птоз, микро-фтальмия. Айрьщша 6enrmepi: саусактарын жабарльщтай жудырыгыныц туюл1 болуы, TeciHiK кыскалыгы, б1рнеше саусагында дога T8pi3fli TepiniK суреттщ болуы. Макалада керсеттген клиникальщ жагдай мысалын-да Эдварде синдромыньщ полиморфтылыгы байкалады: бул балада суйек-булшьщет жуйесшщ тон 93repi-CTepi табылган жок. Наукаска Эдварде синдромы сиякты хромосомальщ ауытку генетикальщ зерттеу нэти-жеа кемег1мен гана койылды.
Туй1нд1 сездер: Эдварде синдромы, 18 трисомия, бала.
SUMMARY
This article discusses the clinical case a girl of 10 months, where there is not a classic manifestation of Edwards syndrome phenotype, causing doubt in the diagnosis. Edwards Syndrome syndrome or trisomy 18 — is a common chromosomal disorder caused by the presence in all cells of the body added hromosomy18. These children are born with intrauterine growth retardation. Clinical symptoms of the disease: gipognotiya expressed delayed mental and motor development, multiple congenital anomalies: the musculoskeletal, cardiovascular, gastrointestinal and central nervous systems. Just marked changes in the organs of vision: corneal clouding membranes, cataract, ptosis, microphthalmia. Distinguishing features: clenched fists with overlapping toes, short sternum, skin pattern in the form of arcs on most fingers. For example, the clinical case described in the article, there is a polymorphism of Edwards syndrome: from the child no changes typical of the musculoskeletal system. Only with the help of genetic research could set the patient chromosomal disorder such as Edwards syndrome.
Edwards Syndrome syndrome or trisomy 18 — is a common chromosomal disorder caused by the presence in all cells of the body added hromosomy18. These children are born with intrauterine growth retardation. Clinical symptoms of the disease: gipognotiya expressed delayed mental and motor development, multiple congenital anomalies: the musculoskeletal, cardiovascular, gastrointestinal and central nervous systems. Just marked changes in the organs of vision: corneal clouding membranes, cataract, ptosis, microphthalmia. Distinguishing features: clenched fists with overlapping toes, short sternum, skin pattern in the form of arcs on most fingers. For example, the clinical case described in the article, there is a polymorphism of Edwards syndrome: from the child no changes typical of the musculoskeletal system. Only with the help of genetic research could set the patient chromosomal disorder such as Edwards syndrome.
Key words: Edward»s syndrome, trisomy 18, child.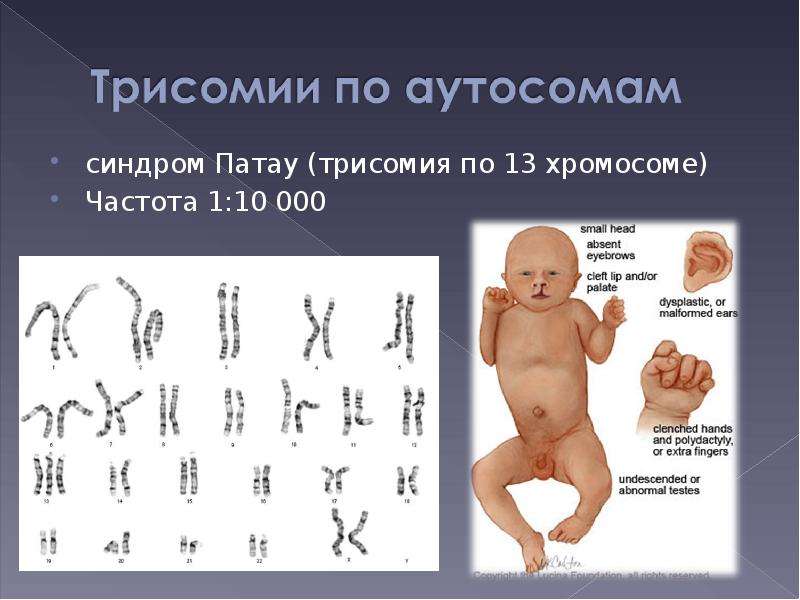
Синдром Эдвардса диагностика трисомии
Синдром Эдвардса – генетическое заболевание, обусловленное трисомией по 18-й паре хромосом. Это довольно редкая аномалия, которая статистически встречается у 1 из 5000 новорожденных. Доказано, что частота формирования данной хромосомной аномалии выше, однако более чем в 70% такая беременность завершается самопроизвольным абортом или мёртворождением.
Проявления синдрома Эдвардса
Так как этот синдром проявляется серьёзными анатомическими аномалиями и патологией внутренних органов, заподозрить его наличие при рождении достаточно легко, как и заподозрить при УЗИ. На этапе беременности синдром проявляется у плода и матери следующими признаками:
● Недостаточная активность эмбриона в утробе матери.
● Малая частота сердечных сокращений плода.
● Слаборазвитая плацента и ее малый размер относительно размеров плода.
● Грыжи брюшной полости.
● Наличие одной пупочной артерии вместо двух.
● Сплетение сосудов, заметное на УЗИ.
● Малый вес плода.
● Асфиксия, выраженное кислородное голодание плода.
Сами по себе эти признаки не показательны и не являются достаточными для постановки диагноза. Достоверно установить синдром Эдвардса возможно только при генетическом исследовании, причем чрезвычайно важную роль играет именно выявление заболевания на ранних сроках беременности. Если при рождении для анализа берут часть тканей новорожденного, то при пренатальной диагностике исследуют плодную ДНК, получаемую либо инвазивным, либо неинвазивным способом
Почему стоит пройти скрининг на синдром Эдвардса
Как и при большинстве других хромосомных патологий, у детей с синдромом Эдвардса велик риск младенческой смертности. В первые 3 месяца умирает более 60% детей, не более трети от общего числа доживает до года. Выжившие страдают различными формами умственной отсталости, анатомическими аномалиями строения скелета и внутренних органов, множественными системными расстройствами, что в большинстве случаев также приводит к ранней смерти.
Относительно благоприятный прогноз возможен лишь при слабовыраженной мозаичной форме заболевания. При надлежащем уходе некоторые дети с ней могут доживать до 10 лет и более. При этом все они страдают серьёзным отставанием в умственном развитии вплоть до имбецильности.
Для устранения физических аномалий, в ряде ситуаций возможна хирургическая коррекция. Основу составляет постоянный уход и медицинское обеспечение, а также относительная симптоматическая коррекция имеющихся нарушений в течение всей последующей жизни. Обычно операции проводят по достижении ребёнком 2-3-летнего возраста – ранее подобные действия нецелесообразны.
Также выжившим и достигшим 3-летнего возраста детям необходима постоянная серьёзная психологическая помощь. В некоторых случаях удается научить элементарным навыкам общения и самообслуживания. Но, к сожалению, результаты такого лечения все-равно сложно считать удовлетворительными, а вероятность даже такого относительно благоприятного исхода очень мала.![]()
В настоящее время не существует способов предотвращения синдрома Эдвардса и других генетических заболеваний. Прерывание беременности на ранних стадиях позволяет сохранить физическое здоровье матери и уберечь ее от серьёзных психологических травм, связанных с последующей младенческой или подростковой смертью ребёнка.
Диагностика синдрома Эдвардса
Как уже говорилось выше, ультразвуковое исследование позволяет лишь косвенно оценить риски синдрома Эдвардса. Для получения более надёжного результата используют пренатальный скрининг, или неинвазивный пренатальный тест — Prenetix. Суть его в исследовании плодной ДНК, присутствующей в крови матери, что даёт при этом синдроме точность результатов на уровне 98%.
К преимуществам теста Prenetix относятся —
● Возможность выявления хромосомных отклонений на ранних стадиях – начиная с 10 недели. Это позволяет выработать специалистам и беременной женщине верное решение в случае неблагоприятного диагноза и подготовиться к прерыванию беременности.
● Высокая чувствительность и специфичность анализа. Определить наличие синдрома Эдвардса с точностью в 98%.
● Неинвазивность забора биоматериала. Взятие крови на исследование может быть проведено в любом процедурном кабинете.
Риск возникновения генетических мутаций у плода растёт с возрастом родителей. Особенно критичен в этом плане возраст матери. Вероятность рождения ребенка с синдромом Эдвардса (а также выкидыша или мёртворождения из-за этого заболевания) достигает 0,7% у женщин с возраста 35 лет и растёт в дальнейшем. Для мужчины критическим считается возраст начиная с 40-45 лет.
Как пройти тест Prenetix?
Чтобы пройти тест Prenetix, необходимо сделать заявку на исследование и сдать кровь. Забор крови на исследование можно произвести в любом регионе страны – в специализированных медицинских учреждениях или лабораториях, имеющих договор с ЦГРМ Genetico. Далее материал будет отправлен на анализ в московскую лабораторию Genetico. Через 12 дней вы сможете получить результат.
Через 12 дней вы сможете получить результат.
Для получения более подробной информации и записи на анализ свяжитесь с нашими специалистами по указанным телефонным номерам или через онлайн-форму на сайте.
Лечение синдрома Эдвардса в Израиле
Синдром Эдвардса относительно распространенное генетическое заболевание, причиной которой является трисомия 18 хромосомы. Родители ребенка больного синдромом Эдвардса – совершенно здоровы по данному заболеванию. Причина патологии – нерасхождение хромосом в процессе созревания половых клеток родителей.
Получить цены в клинике
Обнаружить заболевание можно на ранних стадиях развития плода с помощью забора образца тканей – амниоцентеза а также с помощью тщательного ультразвукового исследования плода, проведенного специалистом.
В медицинском центре Топ Ихилов работают специалисты высшей квалификации, как в области генетического консультирования, так и визуальной диагностики (ультрасаунд). Крайне велика ответственность врача проводящего УЗИ исследование на ранних стадиях развития – упущение может привести к рождению ребенка больного генетическим синдромом.
Заболевание неизлечимо. Частота среди новорожденных – 1 на 7000, заболеваемость среди девочек выше в три раза.
Синдром Эдвардса — тяжелая генетическая патология
Больные страдают от множественных пороков развития – измененная форма черепа, ушных раковин, носа, пороки развития глаз, многочисленные аномалии костно-мышечной системы, крипторхизм, гипоспадия. Интеллектуальное развитие резко замедленно, почти все больные синдромом имеют пороки развития сердечно-сосудистой системы, около половины имеют аномалии системы пищеварения. До года доживают 10% больных.
Беременность протекает со слабой активностью плода, многоводием, отмечается малых размеров плацента, малый вес плода.
Важность пренатальной диагностики
Тяжесть и неизлечимость генетической патологии, вкупе с относительной частотой синдрома обуславливают необходимость тщательных пренатальных проверок, особенно у людей с повышенным риском (возраст старше 30, подверженность вредным экологическим факторам, работа с вредными веществами).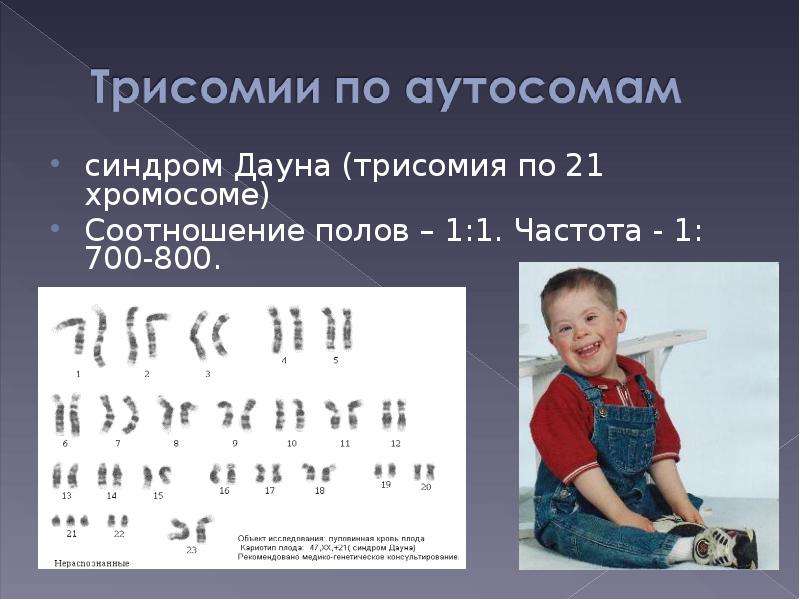
Современная медицина предоставляет возможность пройти десятки генетических тестов на распространенные патологии, а также с помощью высокотехнологичной аппаратуры оценить состояние плода на ранних стадиях развития. Тщательная диагностика позволить вовремя провести аборт по медицинским показаниям и предотвратить рождение ребенка с тяжелыми уродствами развития.
В медицинском центре Топ Ихилов Вы пройдете необходимые обследования и генетическую консультацию специалиста, который поможет Вам спланировать здоровую беременность и родить здорового ребенка.
Получить цены- 5
- 4
- 3
- 2
- 1
Исследование кариотипа (Количественные и структурные аномалии хромосом) с обязательной выдачей кариограммы
Метод определения Культивирование лимфоцитов периферической крови, микроскопия дифференциально окрашенных метафазных хромосом.
Исследуемый материал Цельная кровь (с гепарином, без геля)
Доступен выезд на дом
ИССЛЕДОВАНИЕ НЕ ЯВЛЯЕТСЯ АНАЛОГОМ АНА-ТЕЛОФАЗНОГО МЕТОДА АНАЛИЗА ХРОМОСОМНЫХ АБЕРРАЦИЙ (100 клеток)!
КАРИОТИПИРОВАНИЕ ВХОДИТ В СОСТАВ ИССЛЕДОВАНИЙ: Генетические VIP-профили
Репродуктивное здоровье
Репродуктивное здоровье женщины
Репродуктивное здоровье мужчины
Кариотип — это совокупность признаков полного набора хромосом соматических клеток организма на стадии метафазы (III фаза деления клетки) – их количество, размер, форма, особенности строения. Исследование кариотипа проводят методом световой микроскопии с целью выявления патологии хромосом. Чаще всего это исследование проводят у детей для выявления заболеваний, обусловленных нарушениями в хромосомах и у супругов при бесплодии или привычном невынашивании беременности. Выявление хромосомных перестроек в этом случае позволяет установить причину бесплодия и прогнозировать риск рождения в данной семье детей с хромосомной патологией. Вне процесса деления клетки хромосомы в её ядре расположены в виде «распакованной» молекулы ДНК, и они трудно доступны для осмотра в световом микроскопе. Для того, чтобы хромосомы и их структура стали хорошо видны используют специальные красители, позволяющие выявлять гетерогенные (неоднородные) участки хромосом и проводить их анализ – определять кариотип. Хромосомы в световом микроскопе на стадии метафазы представляют собой молекулы ДНК, упакованные при помощи особых белков в плотные сверхспирализованные палочковидные структуры. Таким образом, большое число хромосом упаковывается в маленький объём и помещается в относительно небольшом объёме ядра клетки.
Исследование кариотипа проводят методом световой микроскопии с целью выявления патологии хромосом. Чаще всего это исследование проводят у детей для выявления заболеваний, обусловленных нарушениями в хромосомах и у супругов при бесплодии или привычном невынашивании беременности. Выявление хромосомных перестроек в этом случае позволяет установить причину бесплодия и прогнозировать риск рождения в данной семье детей с хромосомной патологией. Вне процесса деления клетки хромосомы в её ядре расположены в виде «распакованной» молекулы ДНК, и они трудно доступны для осмотра в световом микроскопе. Для того, чтобы хромосомы и их структура стали хорошо видны используют специальные красители, позволяющие выявлять гетерогенные (неоднородные) участки хромосом и проводить их анализ – определять кариотип. Хромосомы в световом микроскопе на стадии метафазы представляют собой молекулы ДНК, упакованные при помощи особых белков в плотные сверхспирализованные палочковидные структуры. Таким образом, большое число хромосом упаковывается в маленький объём и помещается в относительно небольшом объёме ядра клетки. Расположение хромосом, видимое в микроскопе, фотографируют и из нескольких фотографий собирают систематизированный кариотип — нумерованный набор хромосомных пар гомологичных хромосом. Изображения хромосом при этом ориентируют вертикально, короткими плечами вверх, а их нумерацию производят в порядке убывания размеров. Пару половых хромосом помещают в самом конце изображения набора хромосом. Современные методы кариотипирования обеспечивают детальное обнаружение хромосомных аберраций (внутрихромосомных и межхромосомных перестроек), нарушения порядка расположения фрагментов хромосом — делеции, дупликации, инверсии, транслокации. Такое исследование кариотипа позволяет диагностировать ряд хромосомных заболеваний, вызванных как грубыми нарушениями кариотипов (нарушение числа хромосом), так и нарушением хромосомной структуры или множественностью клеточных кариотипов в организме. Нарушения нормального кариотипа у человека возникают на ранних стадиях развития организма. Если это происходит в половых клеток будущих родителей (в процессе гаметогенеза), то кариотип зиготы (см.
Расположение хромосом, видимое в микроскопе, фотографируют и из нескольких фотографий собирают систематизированный кариотип — нумерованный набор хромосомных пар гомологичных хромосом. Изображения хромосом при этом ориентируют вертикально, короткими плечами вверх, а их нумерацию производят в порядке убывания размеров. Пару половых хромосом помещают в самом конце изображения набора хромосом. Современные методы кариотипирования обеспечивают детальное обнаружение хромосомных аберраций (внутрихромосомных и межхромосомных перестроек), нарушения порядка расположения фрагментов хромосом — делеции, дупликации, инверсии, транслокации. Такое исследование кариотипа позволяет диагностировать ряд хромосомных заболеваний, вызванных как грубыми нарушениями кариотипов (нарушение числа хромосом), так и нарушением хромосомной структуры или множественностью клеточных кариотипов в организме. Нарушения нормального кариотипа у человека возникают на ранних стадиях развития организма. Если это происходит в половых клеток будущих родителей (в процессе гаметогенеза), то кариотип зиготы (см. ), образовавшейся при слиянии родительских клеток, также оказывается нарушенным. При дальнейшем делении такой зиготы все клетки эмбриона и развившегося из него организма окажутся с одинаково аномальным кариотипом. Однако, нарушения кариотипа могут возникнуть и на ранних стадиях дробления зиготы. Развившийся из такой зиготы организм содержит несколько линий клеток (клеточных клонов) с разными кариотипами. Такое многообразие кариотипов во всём организме или только в некоторых его органах называют мозаицизмом. Как правило, нарушения кариотипа у человека сопровождаются различными, в том числе комплексными, пороками развития, и большинство таких аномалий несовместимо с жизнью. Это приводит к самопроизвольным абортам на ранних стадиях беременности. Однако достаточно большое число плодов (~2,5%) с аномальными кариотипами донашивают до окончания беременности. Ниже приведена таблица, в которой представлены заболевания, обусловленные нарушениями в кариотипе.
), образовавшейся при слиянии родительских клеток, также оказывается нарушенным. При дальнейшем делении такой зиготы все клетки эмбриона и развившегося из него организма окажутся с одинаково аномальным кариотипом. Однако, нарушения кариотипа могут возникнуть и на ранних стадиях дробления зиготы. Развившийся из такой зиготы организм содержит несколько линий клеток (клеточных клонов) с разными кариотипами. Такое многообразие кариотипов во всём организме или только в некоторых его органах называют мозаицизмом. Как правило, нарушения кариотипа у человека сопровождаются различными, в том числе комплексными, пороками развития, и большинство таких аномалий несовместимо с жизнью. Это приводит к самопроизвольным абортам на ранних стадиях беременности. Однако достаточно большое число плодов (~2,5%) с аномальными кариотипами донашивают до окончания беременности. Ниже приведена таблица, в которой представлены заболевания, обусловленные нарушениями в кариотипе.
| Кариотипы | Болезнь | Комментарии |
| 47,XXY; 48,XXXY | Синдром Клайнфельтера | Полисомия по X-хромосоме у мужчин |
| 45X0; 45X0/46XX; 45,X/46,XY; 46,X iso (Xq) | Синдром Шерешевского — Тернера | Моносомия по X-хромосоме, в т. ч. и мозаицизм ч. и мозаицизм |
| 47,ХХX; 48,ХХХХ; 49,ХХХХХ | Полисомии по X хромосоме | Наиболее часто — трисомия X |
| 47,ХХ,+21; 47,ХY,+21 | Болезнь Дауна | Трисомия по 21-й хромосоме |
| 47,ХХ,+18; 47,ХY,+18 | Синдром Эдвардса | Трисомия по 18-й хромосоме |
| 47,ХХ,+13; 47,ХY,+13 | Синдром Патау | Трисомия по 13-й хромосоме |
| 46,XX, 5р- | Синдром кошачьего крика | Делеция короткого плеча 5-й хромосомы |
Смотрите также:
Литература
- Фок Р. Генетика эндокринных болезней. — Эндокринология / Под ред. Лавина Н. — М.: Практика, 1999.
- Karger S., Basel. An International System for Human Cytogenetic Nomenclature, Mitelman, F (ed).
 ISCN, 1995.
ISCN, 1995. - Международная классификация болезней. Врождённые аномалии (пороки развития), деформации и хромосомные нарушения (Q00-Q99). Хромосомные аномалии, не классифицированные в других рубриках (Q90-Q99).
- Хромосомные болезни // НЕВРОНЕТ http://www.neuronet.ru/bibliot/semiotika/11_3.html
 ISCN, 1995.
ISCN, 1995.Синдром Эдвардса (трисомия 18) — NHS
Синдром Эдвардса, также известный как трисомия 18, является редким, но серьезным заболеванием.
Синдром Эдвардса влияет на продолжительность жизни ребенка. К сожалению, большинство детей с синдромом Эдвардса умирают до или вскоре после рождения.
Небольшое количество (около 13 из 100) детей, рожденных живыми с синдромом Эдвардса, доживут до своего первого дня рождения.
Причина синдрома Эдвардса
Каждая клетка вашего тела обычно содержит 23 пары хромосом, которые несут гены, унаследованные вами от родителей.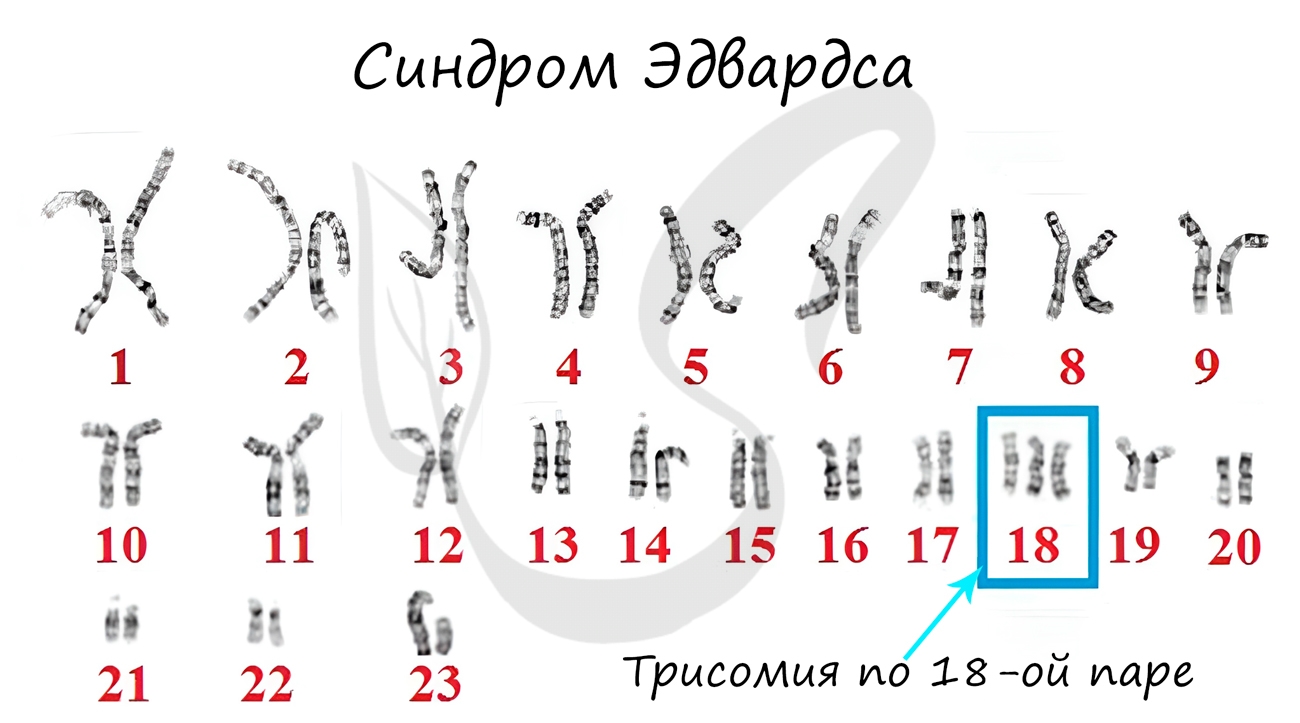
У ребенка с синдромом Эдвардса 3 копии хромосомы номер 18 вместо 2. Это влияет на то, как ребенок растет и развивается. Наличие 3 копий 18 хромосомы обычно происходит случайно из-за изменения сперматозоидов или яйцеклетки до зачатия ребенка.
Ваш шанс родить ребенка с синдромом Эдвардса увеличивается с возрастом, но любой может иметь ребенка с синдромом Эдвардса. Состояние обычно не передается в семье и не вызвано чем-либо, что родители сделали или не сделали.
Обратитесь к терапевту, если хотите узнать больше. Они могут направить вас к генетическому консультанту.
Типы синдрома ЭдвардсаСимптомы и степень серьезности поражения вашего ребенка обычно зависят от того, есть ли у него полный, мозаичный или частичный синдром Эдвардса.
Полный синдром Эдвардса
Большинство детей с синдромом Эдвардса имеют дополнительную 18-ю хромосому, присутствующую во всех клетках. Это называется полным синдромом Эдвардса.
Последствия полного синдрома Эдварда часто бывают более серьезными. К сожалению, большинство детей с этой формой умирают до того, как родятся.
К сожалению, большинство детей с этой формой умирают до того, как родятся.
Мозаика Синдром Эдвардса
Небольшое количество детей с синдромом Эдвардса (примерно 1 из 20) имеют лишнюю 18-ю хромосому лишь в некоторых клетках. Это называется мозаичным синдромом Эдвардса (или иногда мозаичной трисомией 18).
Это может привести к более мягким последствиям заболевания, в зависимости от количества и типа клеток, у которых есть дополнительная хромосома. Большинство рожденных живыми младенцев с этим типом синдрома Эдварда проживут не менее года и могут дожить до 9000 лет.
Частичный синдром Эдвардса
У очень небольшого числа детей с синдромом Эдвардса (примерно 1 из 100) в клетках имеется только часть дополнительной хромосомы 18, а не целая дополнительная хромосома 18. Это называется частичной Синдром Эдвардса (или иногда частичная трисомия 18).
Как частичный синдром Эдвардса влияет на ребенка, зависит от того, какая часть хромосомы 18 присутствует в его клетках.
Консультации для молодых родителей
Есть поддержка для всего, что нужно вам или вашему ребенку.
Все дети, рожденные с синдромом Эдвардса, будут иметь некоторую неспособность к обучению.
Синдром Эдвардса связан с определенными физическими особенностями и проблемами со здоровьем. Каждый ребенок уникален, у него разные проблемы со здоровьем и разные потребности. Обычно они имеют низкий вес при рождении, а также могут иметь широкий спектр физических симптомов. У них также могут быть сердечные, респираторные, почечные или желудочно-кишечные заболевания.
Несмотря на свои сложные потребности, дети с синдромом Эдвардса могут постепенно начать делать больше вещей.
Как и любой ребенок, они:
- обладают собственной индивидуальностью
- учатся в своем собственном темпе
- обладают важными и уникальными для них вещами
Старайтесь не думать слишком далеко и проводить время с ребенком .
Скрининг на синдром Эдвардса
Если вы беременны, вам предложат пройти обследование на синдром Эдвардса на сроке от 10 до 14 недель беременности. Это проверяет вероятность того, что у вашего ребенка это заболевание.
Этот скрининговый тест называется комбинированным тестом. Он определяет вероятность наличия у ребенка синдрома Эдвардса, синдрома Дауна и синдрома Патау.
Во время теста вам сделают анализ крови и ультразвуковое сканирование для измерения жидкости в задней части шеи вашего ребенка (затылочная прозрачность).
Подробнее о скрининге на синдром Эдвардса на 10–14 неделе
Если невозможно измерить жидкость в задней части шеи вашего ребенка, или если вы беременны на сроке более 14 недель, вам предложат скрининг на Синдром Эдвардса как часть вашего 20-недельного сканирования.Иногда это называют сканированием в середине беременности. Это ультразвуковое сканирование, позволяющее определить, как растет ваш ребенок.
Скрининг не может определить, какая форма синдрома Эдвардса может быть у вашего ребенка или как это повлияет на него.
Подробнее о 20-недельном сканировании
Диагностика синдрома Эдвардса во время беременности
Если комбинированный тест показывает, что у вас больше шансов родить ребенка с синдромом Эдвардса, вам будет предложено пройти тест, чтобы точно определить, есть ли у вашего ребенка это заболевание.
Этот диагностический тест включает в себя анализ образца клеток вашего ребенка, чтобы проверить, есть ли у него дополнительная копия хромосомы 18.
Есть 2 разных способа получить этот образец клеток:
Это инвазивные тесты, которые увеличивают ваши шансы выкидыш. Ваш врач обсудит это с вами.
Результаты диагностического теста
Врач-специалист (акушер) или акушерка объяснит, что означают результаты скрининга, и расскажет вам о возможных вариантах.
Это очень сложная ситуация, и нормально испытывать целый ряд эмоций.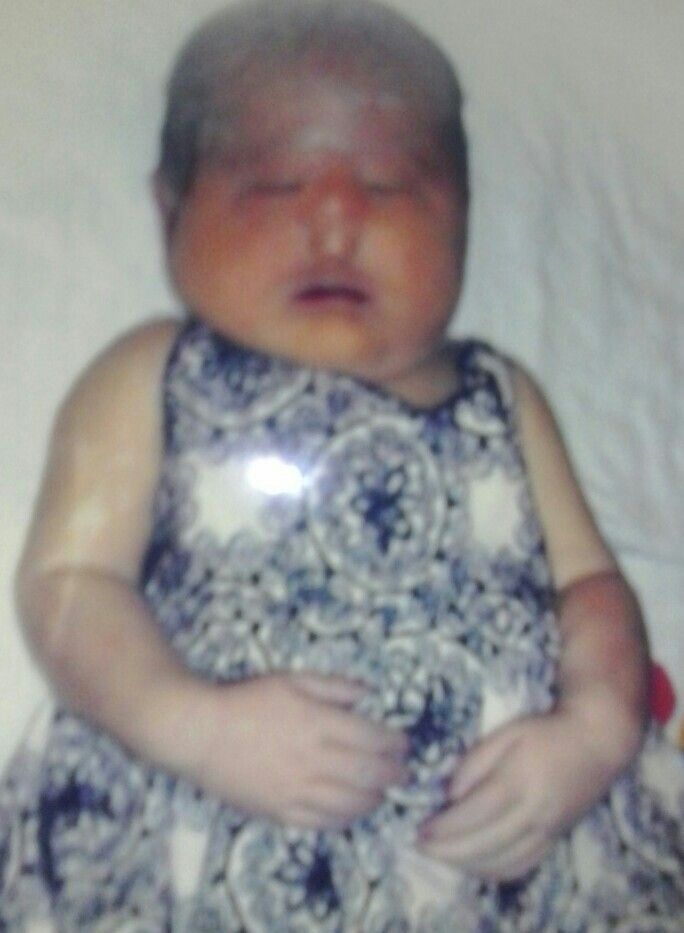 Может быть полезно поговорить со своим врачом, семьей и друзьями или партнером о том, о чем вы думаете и что чувствуете.
Может быть полезно поговорить со своим врачом, семьей и друзьями или партнером о том, о чем вы думаете и что чувствуете.
Если вам сказали, что у вашего ребенка синдром Эдвардса, до или после рождения, вам будет предложена поддержка и информация.
Вы можете посетить веб-сайт SOFT UK, чтобы получить поддержку и дополнительную информацию о синдроме Эдвардса, а также связаться с другими семьями, затронутыми этим заболеванием.
Вы также можете связаться с Antenatal Results and Choices (ARC), где есть информация о скрининговых тестах и о том, что вы можете почувствовать, если вам скажут, что у вашего ребенка есть или может быть проблема.
У ARC есть телефон доверия, на который можно позвонить по телефону 0845077 2290 или 0207713 7486 с мобильного телефона с понедельника по пятницу с 10:00 до 17:30. На горячую линию отвечает обученный персонал, который может предложить информацию и поддержку.
Подробнее о том, что происходит, если дородовые скрининговые тесты что-то обнаруживают
Диагностика синдрома Эдвардса после рождения
Если врачи считают, что у вашего ребенка синдром Эдвардса после рождения, будет взят образец крови, чтобы проверить, есть ли дополнительные копии хромосомы 18.
Лечение синдрома Эдвардса
От синдрома Эдвардса нет лекарства.
Лечение будет сосредоточено на симптомах заболевания, таких как сердечные заболевания, затрудненное дыхание и инфекции. Вашему ребенку также может потребоваться кормление через зонд, так как у него часто возникают трудности с кормлением.
Синдром Эдвардса влияет на движения вашего ребенка, когда он становится старше, и ему может быть полезно поддерживающее лечение, такое как физиотерапия и трудотерапия.
В зависимости от конкретных симптомов вашего ребенка, ему может потребоваться помощь специалиста в больнице или хосписе, или вы сможете ухаживать за ним дома с необходимой поддержкой.
Советы опекунам
Поддержка человека с синдромом Эдвардса может быть одновременно полезным и трудным.
Если вам нужна помощь или вы просто хотите поговорить с кем-нибудь, есть много возможностей поддержки.
В вашем руководстве по социальной помощи и поддержке содержится множество советов о том, как вы можете найти время, чтобы позаботиться о себе, в том числе:
- перерыв в уходе
- получение юридической поддержки и защиты
- забота о своем благополучии
Поговорите с родителями и семьями
Это может помочь поговорить с другими родителями и семьями, которые знают, как вы себя чувствуете.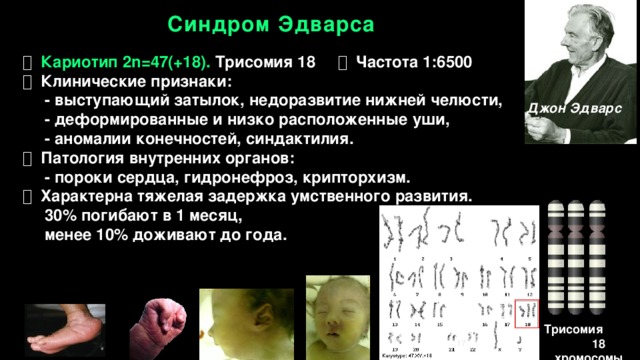
Вы можете сделать это:
- связаться с людьми на форумах и в социальных сетях
- пойти в группу поддержки — спросите у акушерки или патронажного врача о доступных группах поддержки
Информация о вашем ребенке
Если у вашего ребенка будет обнаружен синдром Эдвардса до или после его рождения, его клиническая бригада передаст информацию о нем в Национальную службу регистрации врожденных аномалий и редких заболеваний (NCARDRS).
Это помогает ученым искать более эффективные способы лечения симптомов заболевания. Вы можете отказаться от регистрации в любое время.
Узнайте больше о реестре на GOV.UK
Последняя проверка страницы: 25 сентября 2020 г.
Срок следующей проверки: 25 сентября 2023 г.
Трисомия 18 — NORD (Национальная организация по редким заболеваниям)
Jorde LB, Carey JC и Bamshad MJ. Электронная книга по медицинской генетике. 2015. Elsevier Health Sciences.
2015. Elsevier Health Sciences.
Кэссиди С.Б. и Аллансон Дж. Управление генетическими синдромами. 2010. Джон Уайли и сыновья.
Кэри Дж. Синдром трисомии 18. В: Справочник НОРД по редким заболеваниям. Липпинкотт Уильямс и Уилкинс. Филадельфия, Пенсильвания. 2003: 88.
СТАТЬИ В ЖУРНАЛЕ
Farmakis SG, Barnes AM, et al. Рекомендации по скринингу солидных опухолей при трисомии 18. Am J Med Genet. 2019; 179A: 455-466.
Жанвье А., Фарлоу Б., Баррингтон К.Дж., Бурк С.Дж., Бразг Т. и Уилфонд Б. Укрепление доверия и улучшение общения с родителями детей с трисомией 13 и 18: исследование с использованием смешанных методов.Паллиативная медицина 2019; 0269216319860662.
Като Е., Китасе Ю., Тачибана Т. и др. Факторы, связанные с выживанием при трисомии 18: ретроспективное многоцентровое исследование. Американский журнал медицинской генетики, часть A. 2019; 179: 1253–1259.
Тайра Р., Иноуэ Х, Савано Т. и др. Ведение апноэ у младенцев с трисомией 18. Медицина развития и детская неврология 2019: 25 ноября. Doi: 10.1111 / dmcn.14403. [Epub перед печатью]
Doi: 10.1111 / dmcn.14403. [Epub перед печатью]
Алькхамди М.А., Диого Р. и др. Подробное опорно-двигательный аппарат Изучение плода с трисомией-18 (Эдвардса синдром) с Лангера подмышечной Arch, и сравнение с другими случаями врожденных пороков развития человека.J Anat Sci Res. 2018; 1: 1-8.
Инуое А., Сузуки Р. и др. Терапевтический опыт лечения гепатобластомы, связанной с трисомией 18. Рак крови у детей. 2018; 65: e27093.
Петерсон Р., Каламур Н., Фиоре А., Хаддлстон С. и Спенс К. Факторы, влияющие на исходы после кардиологического вмешательства у младенцев с трисомией 13 и 18. Детская кардиология 2018; 39 (1): 140-147.
Kosiv KA, Gossett JM, Bai S, & Collins RT. Врожденная операция на сердце по поводу госпитальной смертности при трисомии 13 и 18.Педиатрия 2017; 140 (5): e20170772.
Паттерсон Ф. Тейлор Г. и др. Переходы к лечению младенцев с трисомией 13 или 18. Amer J Perinatol. 2017; 34: 887-894.
Петерсон Дж. К., Кочилас Л. К., Каттон К. Г., Моллер Дж. Х. и Сетти С. П.. Отдаленные исходы детей с трисомией 13 и 18 после вмешательств по поводу врожденных пороков сердца. Анналы торакальной хирургии 2017; 103 (6): 1941-1949.
Г., Моллер Дж. Х. и Сетти С. П.. Отдаленные исходы детей с трисомией 13 и 18 после вмешательств по поводу врожденных пороков сердца. Анналы торакальной хирургии 2017; 103 (6): 1941-1949.
Andrews SE, Downey AG и др. Совместное принятие решений и подходы в пренатальном и постнатальном ведении синдромов трисомии 13 и трисомии 18.Am J Med Genet C Semin Med Genet. 2016; 172: 257-263.
Брунс Д.А. и Мартинес А. Анализ пороков сердца и хирургических вмешательств в 84 случаях с полной трисомией 18. Американский журнал медицинской генетики, часть A, 2016 г .; 170 (2): 337-343.
Кэри Дж. С. и Барнс А. М.. Опухоль Вильмса и трисомия 18: есть ли связь? В Американском журнале медицинской генетики, часть C: Семинары по медицинской генетике, 2016 г .; 172 (3): 307-308.
Донован Дж. Х., Кригбаум Дж. И Брунс Д. А. Медицинские вмешательства и выживаемость детей с трисомией 18 по полу.В American Journal of Medical Genetics Part C, Seminars in Medical Genetics 2016; 172 (3): 272-278.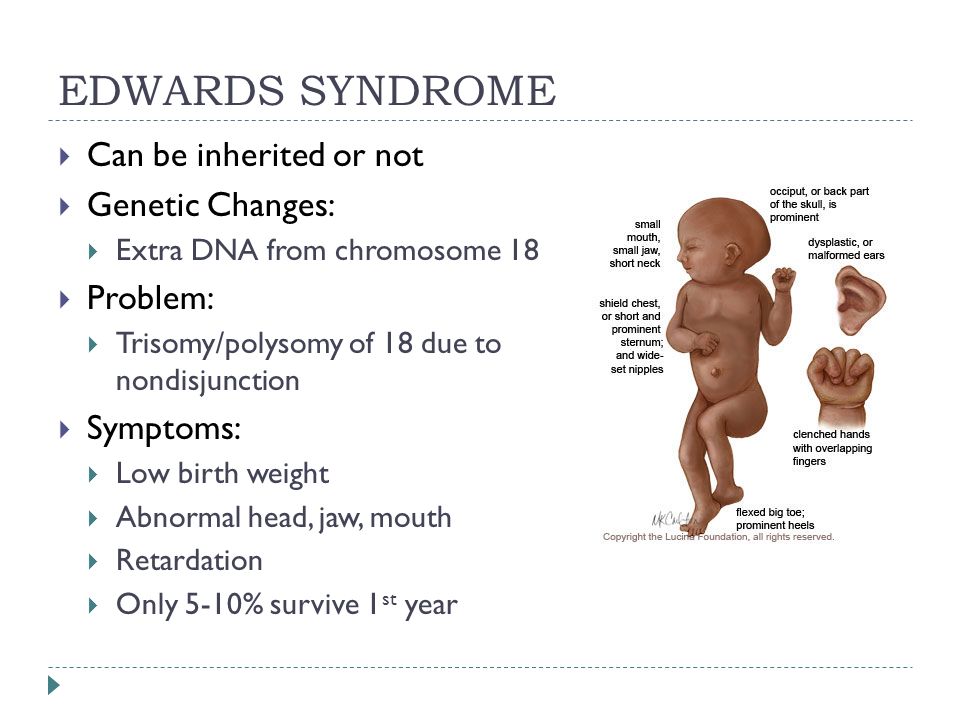
Josephsen JB, Armbrecht ES, Braddock SR, & Cibulskis CC. Процедуры на 1-м году жизни для детей с трисомией 13 и трисомией 18, 25-летний обзор в одном центре. В American Journal of Medical Genetics Part C: Seminars in Medical Genetics 2016 ;. 172 (3): 264-271.
Мейер Р. Э., Лю Дж., Гильбоа С. М. и др. И Национальная сеть профилактики врожденных дефектов. Выживаемость детей с трисомией 13 и трисомией 18: популяционное исследование с участием нескольких штатов.Американский журнал медицинской генетики Part A 2016; 170 (4): 825-837.
Нельсон К.Э., Розелла Л.К. и др. Выживаемость и хирургические вмешательства для детей с трисомией 13 и 18. JAMA. 2016; 316: 420-428.
Сатге Д., Ниши М., Сирвент Н. и Векеманс М. Профиль опухоли при синдроме Эдвардса (трисомия 18). В American Journal of Medical Genetics Part C: Seminars in Medical Genetics 2016; 172 (3): 296-306.
Брунс Д.А. Статус развития 22 детей с трисомией 18 и восьми детей с трисомией 13: значение и рекомендации. Американский журнал медицинской генетики, часть A, 2015 г .; 167 (8): 1807-1815.
Американский журнал медицинской генетики, часть A, 2015 г .; 167 (8): 1807-1815.
Cereda A & Carey JC. Синдром трисомии 18. Журнал редких заболеваний Orphanet 2012; 7 (1): 81.
Palomaki GE, Deciu C, et al. Секвенирование ДНК материнской плазмы надежно идентифицирует трисомию 18 и трисомию 13, а также синдром Дауна: международное совместное исследование. Genet Med. 2012; 14: 296–305.
ИНТЕРНЕТ
Трисомия 18. Информационный центр по генетическим и редким заболеваниям (GARD). Трисомия 18. Последнее обновление: 07.07.2015.https://rarediseases.info.nih.gov/diseases/6321/trisomy-18 По состоянию на 22 января 2020 г.
Что такое трисомия 18? — Trisomy 18 Foundation
Трисомия 18, также известная как синдром Эдвардса, — это состояние, вызванное ошибкой в делении клеток, известной как мейотическая дизъюнкция. Когда это происходит, вместо нормальной пары появляется дополнительная хромосома 18 (тройная) в развивающемся ребенке и существенно нарушает нормальный паттерн развития, что может быть опасно для жизни, даже до рождения. Ошибка трисомии 18 встречается примерно в 1 из каждых 2500 беременностей в Соединенных Штатах и в 1 из 6 000 живорождений. Общее количество родов намного выше, потому что оно включает значительное количество мертворождений, которые происходят во 2-м и 3-м триместрах беременности.
Ошибка трисомии 18 встречается примерно в 1 из каждых 2500 беременностей в Соединенных Штатах и в 1 из 6 000 живорождений. Общее количество родов намного выше, потому что оно включает значительное количество мертворождений, которые происходят во 2-м и 3-м триместрах беременности.
В отличие от синдрома Дауна, который также вызывается лишней хромосомой, проблемы развития, вызванные трисомией 18, связаны с большим количеством медицинских осложнений, которые потенциально опасны для жизни в первые месяцы и годы жизни.Исследования показали, что только 50% доношенных младенцев будут рождены живыми, и у новорожденных девочек будет более высокий процент живорождений, чем у мальчиков.
При рождении госпитализация в отделениях интенсивной терапии новорожденных (ОИТН) является обычным делом для младенцев с трисомией 18. И снова мальчики будут иметь более высокий уровень смертности в этот неонатальный период, чем девочки, хотя дети с более высокой массой тела при рождении чувствуют себя лучше во всех случаях.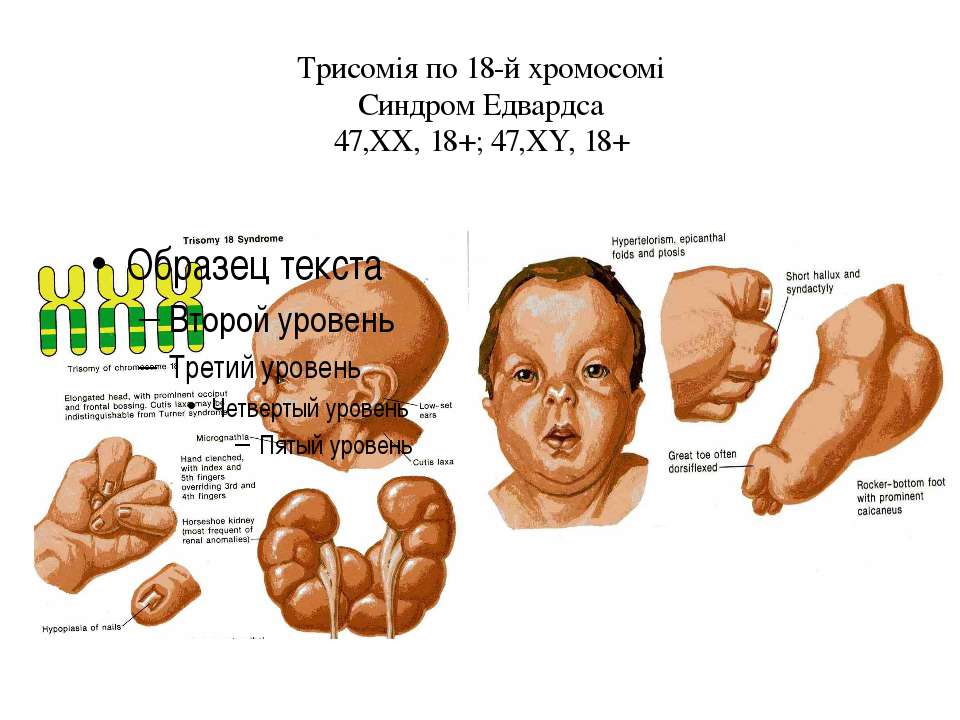 категории.
категории.
Некоторые младенцы смогут выжить до выписки из больницы с помощью ухода на дому для оказания помощи родителям.И хотя 10 или более процентов могут дожить до своих первых дней рождения, есть дети с трисомией 18, которые могут прожить много лет со своими семьями, достигая определенных результатов и участвуя в жизни своего сообщества. Небольшое количество взрослых (обычно девочек) с трисомией 18 доживают и доживают до двадцати и тридцати лет, хотя и со значительными задержками в развитии, которые не позволяют им жить самостоятельно без постоянного ухода.
Дочь Рика Санторума, Белла, имеет трисомию 18
Кандидат в президенты от республиканцев Рик Санторум был вынужден покинуть предвыборную кампанию в минувшие выходные, когда его 3-летняя дочь Изабелла (Белла) была госпитализирована.У Беллы серьезное генетическое заболевание под названием Трисомия 18 , и она была госпитализирована в детскую больницу Вирджинии с пневмонией в обоих легких .
Белла — младшая из семи детей Санторума. При рождении было замечено, что она была «другой», и в пятидневном возрасте Санторуму и его жене Карен поставили диагноз: Трисомия 18 . Им также сказали, что ожидаемая продолжительность жизни детей с трисомией 18 обычно очень коротка и что она, вероятно, не проживет больше года.Несмотря на эти прогнозы, Санторумы сказали врачам, что «мы не собирались концентрироваться на ее смерти, мы собираемся сосредоточиться на ее жизни и сделать все возможное, чтобы помочь ей». Несмотря на частые госпитализации, особенно в течение первого года жизни, Санторум остается оптимистичной:
Некоторые люди описывают таких людей, как Белла, как детей-инвалидов… Я смотрю на радость, простоту и любовь, которые она излучает, и мне ясно, что мы инвалиды. Не она … она права.
Сообщения, поступившие позднее сегодня, показывают, что Белла «свернула за угол», и есть надежда, что ее скоро выпишут домой.
Желаем ей скорейшего выздоровления.
Что такое трисомия?
Давайте пройдемся по переулку памяти и рассмотрим основы генетики:
Мейоз — особый тип деления клеток, необходимый для полового размножения. Клетки, продуцируемые мейозом, называются гаметами , которые у людей относятся к сперматозоидам и яйцеклеткам.
Мейоз отличается от деления клеток «жизненного цикла» митоза двумя важными аспектами:
- Хромосомы в мейозе подвергаются рекомбинации, которая перетасовывает гены, производя различную генетическую комбинацию в каждой гамете, по сравнению с сосуществованием каждой из двух отдельных пар каждой хромосомы (по одной полученной от каждого родителя) в каждой клетке, который возникает в результате митоза.
- Результатом мейоза являются четыре (генетически уникальные) гаплоидные клетки по сравнению с двумя (генетически идентичными) диплоидными клетками, полученными в результате митоза.
Мейоз начинается с одной диплоидной клетки, содержащей две копии каждой хромосомы (всего 46) — одну материнскую и одну отцовскую — и производит четыре гаплоидные клетки, содержащие по одной копии каждой хромосомы (23 хромосомы). Каждая из образующихся хромосом в клетках гамет представляет собой уникальную смесь материнской и отцовской ДНК, гарантируя, что потомство генетически отличается от любого из родителей.
Каждая из образующихся хромосом в клетках гамет представляет собой уникальную смесь материнской и отцовской ДНК, гарантируя, что потомство генетически отличается от любого из родителей.
Нерасхождение — это неспособность пар хромосом правильно разделиться во время стадии 1 или 2 мейоза.Это приводит к клетке с дисбалансом хромосом, и эта клетка называется анеуплоидом .
Если пары хромосом не разделяются должным образом во время деления клетки, яйцеклетка или сперматозоид могут иметь вторую копию одной из хромосом. Если такая гамета приводит к оплодотворению и зарождению эмбриона, полученный эмбрион также может иметь полную копию дополнительной хромосомы.
«Полная трисомия» означает, что была скопирована вся дополнительная хромосома. «Частичная трисомия» означает наличие дополнительной копии части хромосомы.
Трисомия может произойти с любой хромосомой, но часто приводит к выкидышу. Например, трисомия 16 — наиболее распространенная трисомия у людей, встречающаяся более чем в 1% беременностей. Однако это состояние обычно приводит к самопроизвольному выкидышу в первом триместре.
Однако это состояние обычно приводит к самопроизвольному выкидышу в первом триместре.
Из тех трисомий, которые доживают до рождения, наиболее распространенной является Трисомия 21 или Синдром Дауна . Трисомия 18, также известная как синдром Эдвардса , является следующей по частоте встречаемости, за ней следует трисомия 13 или синдром Патау .
Что такое трисомия 18?
Трисомия 18 — относительно частое генетическое заболевание, встречающееся у 1 из каждых 5000 живорождений. У девочек он встречается в три раза чаще, чем у мальчиков.
Большинство случаев трисомии 18 не наследуются, а происходят как случайные события во время мейоза. Это означает, что родителей ничего не сделали до или во время беременности, чтобы вызвать это расстройство у своего ребенка.
Примерно 5% людей с трисомией 18 имеют дополнительную копию хромосомы 18 только в некоторых клетках тела. У этих людей это состояние называется мозаичной трисомией 18. Тяжесть мозаичной трисомии 18 зависит от типа и количества клеток, которые имеют дополнительную хромосому. Развитие людей с этой формой трисомии 18 может варьироваться от нормального до тяжелого.
У этих людей это состояние называется мозаичной трисомией 18. Тяжесть мозаичной трисомии 18 зависит от типа и количества клеток, которые имеют дополнительную хромосому. Развитие людей с этой формой трисомии 18 может варьироваться от нормального до тяжелого.
Очень редко длинное (q) плечо хромосомы 18 присоединяется (перемещается) к другой хромосоме во время образования гамет или на очень ранней стадии эмбрионального развития. У больных людей есть две копии 18-й хромосомы плюс дополнительный материал 18-й хромосомы, прикрепленный к другой хромосоме.Если только часть q-руки присутствует в трех копиях, физические признаки транслокационной трисомии 18 могут отличаться от тех, которые обычно наблюдаются при трисомии 18. Если вся q-рука присутствует в трех копиях, люди могут быть серьезно затронуты, как если бы у них было три полных копии хромосомы 18.
Каковы особенности трисомии 18?
Материал дополнительной хромосомы мешает нормальному развитию. Эти проблемы развития также связаны с медицинскими осложнениями, которые потенциально опасны для жизни в первые месяцы и годы жизни.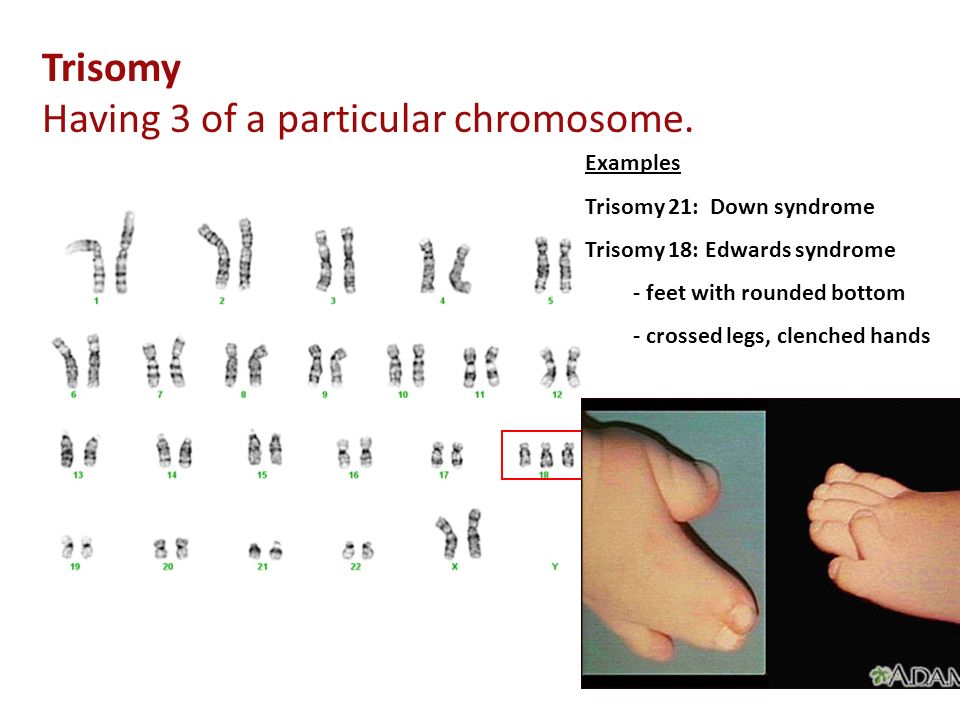 Пятьдесят процентов доношенных младенцев будут мертворожденными. Только 10% детей с трисомией 18 доживают до своего первого дня рождения.
Пятьдесят процентов доношенных младенцев будут мертворожденными. Только 10% детей с трисомией 18 доживают до своего первого дня рождения.Особенности трисомии 18 включают:
- Сжатые руки
- Скрещенные ноги (предпочтительное положение)
- Ножки с закругленным дном (коромысла)
- Низкая масса тела при рождении
- Низко посаженные уши
- Умственная отсталость
- Маленькая голова (микроцефалия)
- Маленькая челюсть (микрогнатия)
- Недоразвитые ногти
- Неопустившееся яичко
- Грудь необычной формы ( pectus carinatum )
Другие признаки включают аномалии глаз, включая колобому радужной оболочки, пупочную или паховую грыжу и диастаз прямых мышц живота.
Часто наблюдаются признаки врожденного порока сердца, наиболее частыми из которых являются дефект межпредсердной перегородки (ДМПП), дефект межжелудочковой перегородки (ДМЖП) или открытый артериальный проток (ОАП).
- Подковообразная почка
- Гидронефроз
- Поликистоз почек
По данным Фонда трисомии 18, «хотя менее 10 процентов доживают до своих первых дней рождения, некоторые дети с трисомией 18 могут прожить много лет со своими семьями, достигая определенных результатов и участвуя в жизни своего сообщества.Небольшое количество взрослых (обычно девочек) с трисомией 18 имеют и доживают до двадцати и тридцати лет, хотя и со значительными задержками в развитии, которые не позволяют им жить самостоятельно без вспомогательного ухода ».
Трисомия 18 — определение, симптомы, изображения, диагностика и ожидаемая продолжительность жизни
«Трисомия 18, также известная как синдром Эдвардса, возникает из-за числовой хромосомной аномалии, при которой дополнительная копия хромосомы 18 присутствует вместе с парой. ”
”
Трисомии — один из распространенных типов числовых хромосомных аномалий, при котором чаще встречаются трисомия 21, трисомия 18 и трисомия 13.
Это распространенный тип генетической аномалии, возникающий из-за неизвестных причин или несбалансированного хромосомного распределения, в отличие от других наследственных мутаций ДНК, трисомия не может быть унаследована от родителей.
Генетические аномалии можно разделить на две категории: генные мутации и хромосомные изменения.
Хромосомные изменения происходят из-за изменений или мутаций в хромосомах, среди них численные хромосомные аномалии и структурные хромосомные аномалии.
Хромосомы — это наследственная единица живого организма, на которой расположены гены. У человека присутствует 23 пары хромосом, каждая хромосома присутствует в паре в каждой клетке, кроме половых клеток.
Парные хромосомы называются диплоидным числом хромосом (2n), таким образом, всего в клетке присутствует 46 хромосом.
При численном изменении хромосомы эти числа меняются, что называется анеуплоидией. Трисомия — это тип анеуплоида, у которого число хромосом увеличивается до 1 (46 + 1 = 47). Изменение ряда хромосом может вызвать серьезные проблемы со здоровьем
В процессе мейоза половых клеток событие, называемое неразъединением, приводит к неравномерному распределению хромосом, вызывая трисомию.
В данной статье мы поговорим об одном из распространенных типов трисомии, известном как синдром Эдварда — трисомии 18.
Содержание статьи:
Трисомия 18, также известная как синдром Эдвардса, представляет собой редкую форму хромосомной аберрации и один из видов врожденных генетических аномалий, вызывающих серьезные проблемы со здоровьем.
В 1960 году об этом впервые сообщил Джон Хилтон Эдвардс, и, следовательно, от его имени он также известен как синдром Эдвардса.
1 из 7000 новорожденных во всем мире страдает трисомией 18. К сожалению, младенцы с синдромом Эдвардса умирают до или сразу после рождения.
Это второй по распространенности тип аутосомной трисомии после синдрома Дауна.
Аутосомная трисомия — трисомия возникает в аутосомной хромосоме и известна как аутосомная трисомия.
Среди 23 пар хромосом 22 пары являются аутосомными хромосомами, тогда как пара половых хромосом определяет пол плода; XX у женщин и XY у мужчин называются половыми хромосомами.
Цитологически состояние трисомии 18 известно как 47 XX / XY, +18
Хотя тяжесть заболевания зависит от типа трисомии 18 и от того, что ребенок может умереть раньше, раньше или позже во взрослом возрасте.Три типа трисомии 18:
Полная трисомия 18:Полная или полная хромосома 18 встречается с парой хромосомы 18. Таким образом, в клетке и во всех клетках присутствуют три различных 18 хромосомы.
Полная трисомия 18 — серьезное врожденное заболевание, ребенок может умереть во время беременности в большинстве случаев или сразу после рождения.
Большинство случаев синдрома Эдварда — полная трисомия 18.
Частичная трисомия 18:Вместо всей хромосомы 18, некоторая часть хромосомы 18 присутствует в клетке с парой.Кроме того, ученым до сих пор неизвестно, какая часть хромосомы 18 частично присутствует в этой паре.
Однако тяжесть заболевания зависит от того, какая дополнительная часть 18 хромосомы присутствует у ребенка. Согласно недавнему исследованию, частичная трисомия 18 может произойти, если у одного из родителей аномальная хромосома 18.
Младенцы с частичной трисомией 18 могут прожить некоторое время или до зрелого возраста со сложными физическими и психическими проблемами.
Мозаичная трисомия 18:В мозаичной ситуации у индивидуума встречаются два разных типа клеточных популяций; один с тремя 18 числами хромосом и один с нормальной парой 18 хромосом.
Отныне тяжесть заболевания зависит от количества клеток с трисомией 18.
У детей с легким поражением меньше пораженных клеток. 10% детей с трисомией 18 имеют мозаичную трисомию и доживают до взрослого возраста.
10% детей с трисомией 18 имеют мозаичную трисомию и доживают до взрослого возраста.
Дополнительная хромосома вызывает серьезные физические и психические проблемы.
Ненаследственная форма генетической условной трисомии 18 возникает из-за события, называемого недизъюнкцией.
Как мы уже говорили выше, все клетки тела имеют 46 хромосом, кроме половых клеток. Он содержит только хромосомы гаплоидного набора; 23 в сперме и 23 в яйцеклетке унаследовали 46 хромосом плоду.
В процессе деления зародышевых клеток — мейоза 1 или мейоза 2 неравномерное распределение хромосом (известное как нерасхождение) вызывает ненормальное распределение хромосом.
Во время второго раунда мейоза (в большинстве случаев) одна из половых клеток вызывает трисомию.Считается, что трисомия происходит от матери, более чем в 90% случаев полная трисомия.
Трисомия 18 вызвана процессом нерасхождения неравномерного хромосомного распределения.
«Трисомия 18 — это тип хромосомной аберрации, при которой дополнительная копия хромосомы 18 возникает вместе с парой в процессе нерасхождения».
На трисомии 18 показан широкий спектр психических и физических симптомов. Вот некоторые из общих симптомов:
- Низкий вес при рождении
- Сравнительно маленькая челюсть и рот
- Аномальная и меньшая головка
- Ухо низко посаженное
- Расщелина неба
- Слаборазвитые большие пальцы рук со сжатыми стопами
- Проблемы с дыханием
- Сердце, почки, а также проблемы с другими органами
- Инфекции мочевыводящих путей (частые)
- Обучаемость и умственные отклонения
- Изогнутый корешок
- Малый корпус
- Умственные и психические отклонения
Примечание: в Индии зарегистрировано около 100 000 случаев трисомии.
Фотография ребенка с трисомией 18; (A) аномальные пальцы, (B) сжатые стопы (C) недоразвитые руки и пальцы.
Некоторые из общих физических характеристик настоящего состояния включают когнитивные и психомоторные нарушения, дефицит роста, черепно-лицевые особенности, преобладающую фигуру и характерную осанку рук, гипоплазию ногтей, короткую грудину и большой палец стопы.
Новорожденный ребенок с трисомией 18 с гранулами расщелины.
Беременности с трисомией 18 подвержены высокому риску мертворождения, потери плода и гибели плода.
Ультразвук и кариотипирование — тип генетического теста обычно используется в качестве диагностического инструмента для скрининга трисомии 18, при котором кариотипирование используется для проверки результатов ультразвукового исследования.
В дополнение к этому, генетический тест нового возраста теперь также используется для скрининга текущего состояния, которое мы обсудим в этой части позже.
УЗИ:Ультразвуковой метод используется в качестве основного метода скрининга на любой тип трисомии, независимо от ее показаний, позже подтвержденный генетическим тестированием.
На 10–14 неделе беременности УЗИ можно использовать для первичного скрининга.
Если может быть обнаружен какой-либо риск, связанный с беременностью, беременные немедленно направляют на генетическое тестирование.
Кариотипирование:Часто известный как хромосомный анализ — метод генетического тестирования, отличный от генного тестирования.
Здесь сначала у плода берут образец околоплодных вод (амниоцентез) или ворсинок хориона и отправляют в генетическую лабораторию.
Профессиональный генетик выращивает метафазные клетки для получения метафазных хромосом, а затем выполняет кариотипирование, чтобы найти наши генетические аберрации, связанные с хромосомами.
При использовании техники полос G или Giemsa можно встретить хромосомные аберрации, если таковые имеются.
Кариограмма составляется с помощью компьютерного программного обеспечения, которое помогает найти связанные с ней числовые или структурные хромосомные аномалии.
Типичная кариограмма пациента с трисомией 18, выполненная с использованием G-бэнда.
Помимо трисомии 18, с помощью этого метода также можно встретить другие трисомии, такие как трисомия 21 или трисомия 13.
Хотя этот метод является болезненно-инвазивным, и вероятность или риск потери беременности у него выше.
Прочтите нашу статью «Кариотипирование: протокол кариотипирования для культуры лимфоцитов периферической крови».
Неинвазивная пренатальная диагностика:В настоящее время ученые используют неинвазивный метод для скрининга трисомии 18, известный как НИПД, часто известный как тестирование внеклеточной ДНК плода.
Во время беременности некоторое количество ДНК плода также циркулирует в крови матери, называемой внеклеточной ДНК.
Из крови матери выделяется ДНК плода, с помощью которой проводится генетический тест на выявление трисомии 18. Обычно для этого используется количественная ПЦР.
ДНК плода очень мало присутствует в материнской крови, поэтому иногда она не может дать результатов, хотя это неинвазивно.
Отныне, в отличие от обычного кариотипирования, он не может часто практиковаться.Для использования в рутинной диагностике необходимы дополнительные исследования, протоколы и валидация.
Позже, во время беременности, на 21 неделе беременности, для подтверждения результатов может быть проведено сканирование на предмет выявления врожденных аномалий.
Средняя продолжительность жизни детей с трисомией 18 или полной трисомией 18 составляет от 2 дней до 2 недель. Большинство младенцев умирают в утробе матери или сразу после рождения.
Младенцы с мозаичной или частичной трисомией могут дожить до взрослого возраста. До 80% детей с синдромом Эдвардса умирают в течение 21 часа.
5–10% младенцев с трисомией 18 могут дожить до 1 года.
Интересно, что примерно 80% случаев трисомии 18 происходит у женщин, мы можем сказать, что беременность женским плодом более подвержена риску трисомии 18.
Синдром Эдварда часто встречается у младенцев женского пола, однако уровень внутриутробной смертности выше у плода мужского пола.
Никто не может принуждать кого-либо к прерыванию беременности, это решение родителей, особенно решение матери.С этим связано множество социальных и эмоциональных проблем, поэтому мы можем посоветовать паре, что делать дальше, если у беременных выявлена трисомия 18.
Прежде всего, не поддавайтесь панике и эмоциям, сразу же обратитесь к врачу.
Сохраняйте спокойствие и объясните ситуацию своему врачу и поймите, что они говорят вам.
Очень меньше шансов, что ребенок с полной трисомией 18 выживет даже после рождения.
, даже если это возможно, родители должны страдать эмоционально, экономически и социально; никто не хочет видеть, как их ребенок страдает от боли.
Итак, если ваш врач или генетический консультант посоветовал сделать аборт; спокойно подумай об этом.
Это ненаследственное состояние и полностью случайное, до тех пор, пока у кого-то из вас нет хромосомных аномалий, связанных с хромосомой 18.
Никто из вас (ни вы, ни часовой муж) не несет за это ответственности, поэтому не вините друг друга в этом и разбирайтесь в ситуации.
Даже если один из вас не хочет прерывать его, уважайте решение друг друга и продолжайте.
Помните, вероятность возникновения трисомии 18 увеличивается с увеличением возраста матери. Женщины старше 30 лет относятся к группе высокого риска трисомии 18.
Нет таких методов лечения трисомии 18, поэтому необходимы программы пренатального скрининга для предотвращения генетических аномалий, таких как трисомия 18. Трисомия 18 не следует какому-либо конкретному образцу наследования, поэтому вероятность ее унаследования при следующей беременности незначительна.
Ресурсы:
Cereda A, Carey JC.Синдром трисомии 18. Орфанет J Редкий Диск . 2012; 7: 81. Опубликовано 23 октября 2012 г.
Трисомия 18
Трисомия 18 — это генетическое хромосомное заболевание, характеризующееся наличием дополнительной хромосомы, вызывающей аномалии в различных частях тела. Также известный как синдром Эдвардса, это вторая по распространенности трисомия после трисомии 21 (синдром Дауна).
Трисомия 18 возникает, когда хромосомные клетки не могут должным образом делиться во время беременности.Обычно хромосомы, из которых состоит ваше тело, состоят из двух наборов, а здоровый геном состоит из 23 пар. В случае трисомии 18 у человека развивается третья хромосома в 18-й паре. Дополнительная хромосома, которая будет присутствовать в каждой клетке тела, вызывает серьезные и часто фатальные врожденные дефекты.
Возможны три типа трисомии:
- Полная трисомия 18: Самый распространенный вариант состояния (около 95% всех случаев). В случае полной трисомии 18 дополнительная хромосома обнаруживается в каждой клетке тела ребенка.Эта форма не является наследственной.
- Частичная трисомия 18: Очень редкая форма трисомии 18, которая возникает, когда лишняя хромосома лишь частично присутствует в клетках. Это происходит, когда часть 18 хромосомы прикрепляется к другой хромосоме, что может произойти до или после зачатия. В этом случае синдром может быть вызван наследственными факторами.
- Мозаичная трисомия 18: Мозаичная трисомия также очень редко возникает, когда дополнительная хромосома 18 присутствует только в некоторых клетках организма, а не во всех.Подобно полной трисомии 18, мозаичная трисомия возникает случайно во время деления клеток и не передается по наследству от родителей.
Трисомия 18 обычно диагностируется врачом во время беременности, другими словами, когда ребенок еще находится в утробе матери. Тем не менее, процесс тестирования на это заболевание остается одинаковым независимо от того, проводится ли он до или после рождения. Чтобы проверить наличие трисомии 18, образец ДНК извлекается либо из крови ребенка, либо из тканей тела и культивируется, чтобы врач мог изучить картину хромосом, известную как кариотип.
Для успешного получения кариотипа ДНК ребенка хромосомы выделяют, окрашивают и затем исследуют под микроскопом, который затем может сделать фотографию для исследования. Этот кариотип должен показать вашему врачу, действительно ли в клетках вашего ребенка присутствует третья, дополнительная хромосома 18.
Другой распространенный метод тестирования на трисомию 18 и другие типы аномалий — это скрининг материнской сыворотки, также известный как тест с множественными маркерами. Этот пренатальный скрининг проводится либо в течение первого триместра (Комбинированный скрининг в первом триместре, или «CFTS»), либо во втором триместре (Скрининг материнской сыворотки во втором триместре, или «2TMSS»).
В ходе этих обследований измеряется несколько веществ в крови беременной женщины. Имея эту информацию, врачи могут оценить вероятность того, что на беременность повлияет хромосомное заболевание или некоторые другие проблемы развития, разделив женщин на категории «с высокой вероятностью» или «с низкой вероятностью».
Как диагностируется трисомия 18 во время беременности?Трисомию 18 часто подозревают во время беременности на основании анализа крови или ультразвукового исследования. Но анализ крови или УЗИ — это не путь к уверенному диагнозу.Существует также неинвазивный пренатальный тест (НИПТ), который расскажет вам, обычно в течение 10 дней, гораздо больше о вашем относительном риске рождения ребенка с синдромом Дауна или трисомией 18. Но даже этот тест не является окончательным. Единственный окончательный тест — это амниоцентез. Обратной стороной этого теста является 1 шанс из 200 выкидыша из-за теста, который включает в себя введение тонкой иглы в живот для извлечения околоплодных вод.
Какова продолжительность жизни при трисомии 18?Трудно говорить о продолжительности жизни с трисомией 18.Это просто очень грустно. Большинство детей не доживают до первого месяца. Среди младенцев, переживших первый месяц жизни, 36% выжили в возрасте 1. Только десять процентов детей с трисомией 18 доживают до 10 лет.
Что такое синдром Эдвардса?Синдром Эдвардса — еще одно название трисомии 18.
Является ли трисомия 18 наследственной?Большинство случаев трисомии 18 не передаются по наследству. В этом никто не виноват. Однако родители, у которых есть ребенок с трисомией 18, имеют больший риск иметь еще одного ребенка с этой хромосомной аномалией.
Каковы симптомы трисомии 18?Как и синдром Дауна, эффекты трисомии 18 могут варьироваться от легких до тяжелых, что означает, что она будет проявляться у каждого ребенка по-разному. Независимо от того, насколько легкой или тяжелой может быть случай, наличие трисомии 18 вызывает множество проблем.
Общие проблемы, связанные с трисомией 18, включают:
- Пороки сердца
- Дефект межжелудочковой перегородки (VSD): отверстие между нижними камерами
- Дефект межпредсердной перегородки (ASD): отверстие между верхними камерами
- Коарктация межжелудочковой перегородки аорта: суженный выходной сосуд из сердца
- Дефекты почек
- Омфалоцеле: когда часть кишечного тракта выходит за пределы желудка
- Атрезия пищевода: когда пищевод и желудок не соединяются
- Полигидрамнион: избыток количество околоплодных вод
- Кисты сосудистого сплетения: очаги жидкости в головном мозге
- Микрогнатия (маленькая челюсть) и микроцефалия (маленькая голова)
- Spina bifida: спинной мозг, который не полностью сформирован
- Тонкое, хрупкое и маленькое тело размер, даже при доношенных сроках
- Черепно-лицевые аномалии: аномалии челюсти, черепа, ушей и шеи)
- Ступни с «коромыслом»: ноги и d стопы застряли в изогнутом положении
- Неспособность полностью развести пальцы
- Укороченная грудина
К сожалению, исследования показывают, что от этого состояния связан очень высокий уровень смертности.Средняя продолжительность жизни младенцев, рожденных с этим заболеванием, составляет от 3 дней до 2 недель. Исследования показали, что шанс выжить в первый год составляет 5-10%.
В связи с этим большая часть ухода за младенцами, рожденными с синдромом Эдвардса, в большей степени сосредоточена на паллиативной помощи и оптимизации качества жизни. Лечение осложнений зависит от пораженного органа и тяжести состояния. Лечение синдрома Эдварда будет полностью зависеть от тяжести симптомов, имеющихся у ребенка.
ЗаключениеК сожалению, диагноз трисомии 18 означает, что вам, как родителю, придется принимать чрезвычайно трудные решения относительно ухода за своим ребенком. Есть много ресурсов, которые могут помочь вам в этом процессе, от интервенционных служб, хосписов, социальных работников и генетических консультантов. Также существует множество групп поддержки, созданных для семей, которые сталкиваются с теми же проблемами, которые являются отличным источником поддержки в вашей работе, чтобы справиться с этим диагнозом.
Дополнительная литература- «Синдром трисомии 18» Анны Середа и Джона К. Кэри, Журнал редких заболеваний Orphanet , 2012.
Обзор синдрома трисомии 18, его причин и симптомов. № - «Использование ориентированного на пациента ухода после пренатальной диагностики трисомии 18 или трисомии 13», Шелли Хауг, доктор медицины и др. JAMA Pediatr , 2017.
Исследование эффективности ухода за пациентом (PCC) в случаях пренатальной диагностики трисомии 18 в соответствии с рекомендациями Института медицины для улучшения здравоохранения в США.Подход PCC может снизить вред, когда неадекватные стратегии ухода и консультирования создают конфликт между родителями и опекунами по поводу лечения младенцев с трисомией 18. - «Комбинированный тест первого триместра и интегрированные тесты для скрининга синдрома Дауна и трисомии 18» Гералин М. Мессерлиан, доктор философии и др., 2019.
Различные тесты на трисомию 18 и другие типы генетических аномалий, которые доступны новым матерям в первом и втором триместрах беременности. - «Длительная выживаемость пациента с трисомией 18 и синдромом Денди-Уокера» Феррейра де Соуза LM et al., Medicina , 2019.
Исследование пациента с диагнозом полной трисомии 18, синдромом Денди-Уокера и врожденным пороки сердца на долгосрочную выживаемость. Пациентка — 12-летняя девочка, перенесшая кардиохирургическую операцию в 1 год и 7 месяцев, что позволяет предположить, что раннее хирургическое вмешательство и многопрофильное наблюдение могут изменить клинический исход состояния с летальным исходом.Однако необходимы дальнейшие исследования, чтобы правильно оценить пользу инвазивных процедур, таких как кардиохирургия. - «Врожденная хирургия сердца в отношении госпитальной смертности при трисомии 18», Кэтрин А. Косив и др., Журнал Американской академии педиатрии , 2017.
Исследование врожденного порока сердца (ВПС), распространенного симптома трисомии 18 (t18), чтобы определить влияние хирургии врожденных пороков сердца (CHS) на внутрибольничную смертность. Результаты показали, что ИБС присутствовала у 91% младенцев с диагнозом t18 и у 81% младенцев с диагнозом трисомия 13 (t13).Было показано, что внутрибольничная летальность снизилась у пациентов, перенесших CHS (на 64% ниже у t18 пациентов), и оставалась таковой в следующие 24 месяца наблюдения. Эти результаты позволяют предположить, что в некоторых случаях CHS может быть полезным.
Диагностика и прогноз трисомии синдрома Эдвардса 18
Одним из наиболее серьезных заболеваний, проверяемых при обычном пренатальном диагностическом обследовании, является синдром Эдвардса, хромосомное заболевание с ужасным прогнозом. Синдром Эдвардса также известен как синдром трисомии 18, потому что существует три копии хромосомы 18.
Статистика
Синдром Эдвардса встречается у 1 из 4 000 рождений. Однако заболеваемость во время беременности намного выше, потому что она может быть нераспознанной. У многих женщин с трисомией 18 у плода случается выкидыш или мертворождение.
Прогноз
К сожалению, результаты у детей, рожденных с синдромом Эдвардса, неблагоприятны, в том числе:
- Половина младенцев не выживает дольше первой недели жизни.
- Около 90–95% детей умрут до своего первого дня рождения.
- Некоторые дети доживают до подросткового возраста, но имеют серьезные проблемы со здоровьем и развитием.
Типы
Наиболее распространенной формой синдрома Эдвардса является полная трисомия 18, то есть у ребенка есть три полные копии 18-й хромосомы вместо двух. Также возможна частичная трисомия 18, при которой есть две полные копии хромосомы 18, а также дополнительная частичная копия. Еще один тип — мозаичная трисомия 18, что означает воздействие на некоторые, но не на все клетки.
Последние два типа редки по сравнению с полной трисомией 18. Пациенты с мозаичной трисомией 18 могут быть серьезно затронуты или нет. Фактически, у пациента может быть мозаичная трисомия 18 и он не знает об этом, пока у него или нее не будет диагностирована трисомия 18 у ребенка.
Причины и факторы риска
Дополнительная копия хромосомы 18 уже присутствует во время оплодотворения и является результатом случайных ошибок при делении клеток. Трисомия 18 может возникнуть у родителей всех возрастных групп, но риск наиболее высок, когда матери старше 35 лет.Хотя большая часть трисомии 18 возникает из-за случайных ошибок, частичная трисомия 18 может быть унаследована.
Диагностика
Амниоцентез, взятие проб ворсинок хориона или скрининг в первом триместре с анализом крови и ультразвуковой оценкой воротниковой складки могут помочь в диагностике синдрома Эдвардса. Трисомия 18 обычно диагностируется до рождения ребенка на основании возраста матери, анализов крови и / или признаков отклонений на сонограмме.
Риск повторения
В большинстве случаев трисомия 18 — случайное явление из-за проблем с делением клеток.В редких случаях родители являются носителями частичной трисомии 18 из-за состояния, называемого сбалансированной транслокацией, которое увеличивает риск будущих беременностей.
Если есть вероятность, что вы являетесь носителем, ваш врач может направить вас к генетическому консультанту, чтобы обсудить возможные варианты. Но большинство родителей, у которых есть дети с трисомией 18, не являются носителями.
Диагноз синдрома Эдвардса
Диагноз синдрома Эдвардса — ужасная новость. Многие родители предпочитают прервать беременность после получения подтверждения, что у ребенка определенно есть трисомия 18, учитывая высокий риск серьезных проблем со здоровьем и низкие шансы ребенка выжить в младенчестве.
Другие все равно решают продолжить беременность либо из-за убеждений против абортов, либо из-за того, что они хотят ценить время с ребенком, даже если оно короткое. Нет «правильного» выбора, что делать в этой ситуации. Родители, столкнувшиеся с этим диагнозом, должны делать то, что им кажется правильным.
.